Application note
How Reactive Is Your Warhead? Ranking Covalent Electrophiles
Covalent inhibitors Reactivity Drug design
Some of the most important drugs of the last decade don't just bind their target — they weld themselves to it. Osimertinib (EGFR), ibrutinib (BTK), and the KRAS G12C blockbusters sotorasib and adagrasib all form a permanent covalent bond from a small reactive group — a warhead — to a cysteine on the protein. The whole class lives or dies on one design decision: how reactive should that warhead be?
Too tame, and the drug never engages its target. Too hot, and it labels every cysteine in the proteome — a recipe for off-target toxicity. The sweet spot is narrow, and historically it has been found the slow way: synthesize a series, measure rate constants, iterate. This post shows how a quantum-chemical descriptor turns that into a first-principles triage you can run in minutes, before a single flask comes out — and how it lands the warhead that actually dominates approved covalent drugs.
What the warhead reaction really is
Targeted covalent inhibitors (TCIs) almost all use an acrylamide or a close relative as the warhead, and they react by a textbook Michael addition. The target cysteine, deprotonated to its nucleophilic thiolate, attacks the warhead's electron-poor β-carbon — the far end of the C=C–C=O system. In molecular-orbital language, the thiolate's lone pair flows into the warhead's LUMO: the π* antibonding orbital of the conjugated acceptor. The lower that LUMO sits, the more eagerly it accepts electron density — the more electrophilic the warhead.
That gives us a clean, calculable handle on intrinsic reactivity. Two descriptors capture it. The first is simply the LUMO energy. The second is the Parr electrophilicity index ω, which folds together how much a molecule wants electrons and how hard it resists holding them:
omega = mu^2 / (2 * eta)
mu = (eHOMO + eLUMO) / 2 (electronic chemical potential)
eta = eLUMO - eHOMO (chemical hardness, the HOMO-LUMO gap)A low-lying, accessible LUMO drives ω up; a soft, easily-polarized acceptor pushes it up further. Both numbers fall straight out of a single ground-state calculation, so Hilbeon can rank an entire congeneric series the moment the structures exist.
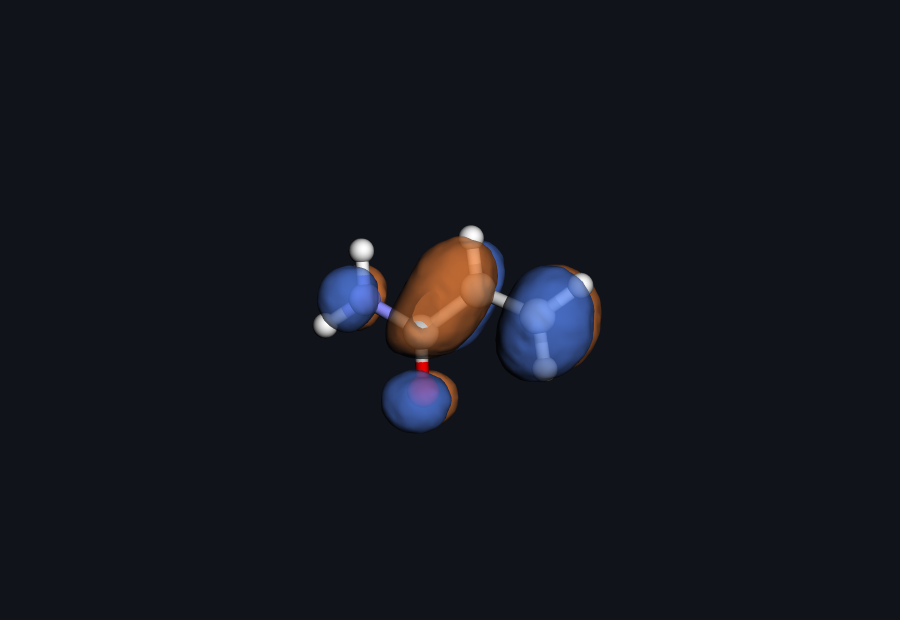 Acrylamide LUMO — drag to rotate
Acrylamide LUMO — drag to rotateFigure: the acrylamide LUMO computed by Hilbeon (B3LYP/6-31G(d), cube file). This π* is literally the orbital the cysteine thiolate attacks in the Michael addition. Blue and orange lobes are the two signs of the wavefunction; notice how the density piles up across the C=C–C=O acceptor.
That is not a textbook cartoon — it is the real wavefunction Hilbeon wrote to a cube file. When a medicinal chemist tunes a warhead, this is the orbital they are reshaping, whether they think in those terms or not.
Ranking six warheads
We took six Michael-acceptor warheads spanning the chemistry actually used in covalent design — nitrile, ketone, ester, and three amide variants — and ran them all through the same Hilbeon calculation. The result is a single ranked list of intrinsic electrophilicity.
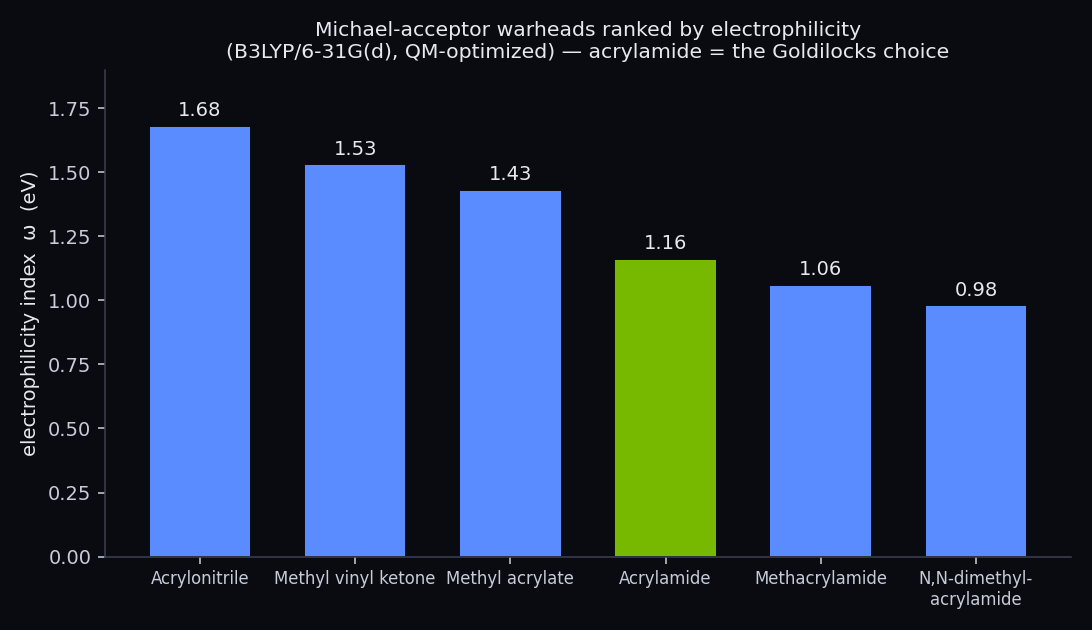
Six Michael-acceptor warheads ranked by the electrophilicity index ω (B3LYP/6-31G(d), Hilbeon). Acrylamide sits in the middle.
| Warhead | LUMO (eV) | ω (eV) |
|---|---|---|
| Acrylonitrile | −1.44 | 1.68 |
| Methyl vinyl ketone | −1.39 | 1.53 |
| Methyl acrylate | −1.10 | 1.43 |
| Acrylamide | −0.74 | 1.16 |
| Methacrylamide | −0.57 | 1.06 |
| N,N-dimethylacrylamide | −0.47 | 0.98 |
Read top to bottom, ω falls from 1.68 eV for acrylonitrile to 0.98 eV for N,N-dimethylacrylamide — a 1.7-fold spread in intrinsic electrophilicity across the series. The LUMO climbs in step — with one near-tie inversion at the very top (see note) — from −1.44 eV to −0.30 eV. Two independent descriptors, one consistent story.
Rank your own warhead series
From a list of SMILES to a ranked electrophilicity table in a couple of prompts. See where your warheads land before you make them.
The reveal: the descriptor predicts the design choice
The ranking is not just internally consistent — it reproduces medicinal-chemistry intuition almost line for line. The esters, ketones and nitriles are the hot electrophiles, clustered at the top. The amides are milder, because the nitrogen lone pair donates into the carbonyl and raises the π*. Add an α-methyl group (methacrylamide) and you cool it further; cap the nitrogen with two methyls (N,N-dimethylacrylamide) and it drops to the bottom of the list. Every substituent effect that a chemist would predict by hand shows up in the numbers, in the right order.
And here is the punchline. Acrylamide sits squarely in the Goldilocks middle of the ranking — ω = 1.16 eV, LUMO = −0.76 eV — reactive enough to engage a target cysteine, tame enough to leave the rest of the proteome alone. That is precisely the balance approved covalent drugs are tuned to, which is why an acrylamide (or substituted-acrylamide) warhead sits in osimertinib, ibrutinib, sotorasib and adagrasib. The descriptor, computed from nothing but the electronic structure, lands on the exact group the field converged on empirically over years of medicinal chemistry.
A note on the top of the list. ω and the LUMO energy agree on the overall trend, but they need not agree on every adjacent pair. Here the two hottest warheads — acrylonitrile and methyl vinyl ketone — are separated by only 0.05 eV in ω, and the LUMO actually orders them the other way. Treat the very top of the ranking as a tie within method noise; the useful signal is the coarse ordering across chemotypes, not a hair-splitting rank between two near neighbours.
What this descriptor is — and is not. ω and the LUMO energy are an intrinsic-reactivity triage for ranking congeneric warheads. They are not an absolute rate constant and not a binding affinity. Real cysteine labeling also depends on the cysteine's pKa (whether the thiolate is even formed), the pocket microenvironment, sterics and orientation, and the actual addition barrier ΔG‡ — a natural refine target is to compute the thiolate-addition transition state explicitly. Level of theory throughout: B3LYP/6-31G(d), gas phase, on QM-optimized geometries (RDKit ETKDG+MMFF starting structures, then geometry-optimized by Hilbeon; the starting and optimized structures differ by only ~0.02–0.3 Å RMSD for these small, rigid acceptors).
How to do it yourself
Every number above came from talking to the Hilbeon assistant in plain language — no input decks, no basis-set syntax to memorize:
build acrylamide from SMILES C=CC(=O)N
run B3LYP/6-31G(d)
report the HOMO and LUMO energies and the electrophilicity index
render the LUMO cube
repeat for acrylonitrile, methyl vinyl ketone, methyl acrylate,
methacrylamide and N,N-dimethylacrylamide, then rank by omegaPrefer scripts? Every one of those steps is also a call in Hilbeon's MCP / HTTP API, so screening a whole warhead library is a loop in a notebook or a CI job. Hilbeon runs on its own GPU-accelerated integral engine (McMurchie–Davidson + Rys — no third-party libraries), fully deterministic and reproducible to the last digit across machines, so the orbitals you are ranking are the real ones.
The takeaway
A covalent drug is a tuning problem, and the tuning knob is the warhead's LUMO. Map that knob with the Parr electrophilicity index and you can rank a whole series from first principles in minutes — esters, ketones and nitriles run hot; amides run mild; α-methyl and N,N-dialkyl substitution cool them further. Acrylamide lands in the middle of the range, which is exactly why it dominates the approved TCIs. The descriptor is a triage, not a verdict: use it to prioritize which warheads to make, then nail the regiochemistry and the rate with an explicit transition-state calculation. Point the same engine at your own series and find the Goldilocks warhead before you synthesize the wrong one.
Rank your own warhead series
Start a 30-day guided pilot — every method, every core, your GPU — and triage the electrophilicity of the warheads on your bench before you make them.
References
- Parr electrophilicity index — R. G. Parr, L. von Szentpály & S. Liu, J. Am. Chem. Soc. 1999, 121, 1922
- Targeted covalent inhibitors, a review — Singh, Petter, Baillie & Whitty, Nat. Rev. Drug Discov. 2011
- Acrylamide-warhead drugs — osimertinib, ibrutinib, sotorasib
- Where the body attacks a molecule — Where Will the Body Attack Your Molecule?