Application note
A Computational Portrait of Aspirin
Drug discovery DFT pKa ESP NMR
You know aspirin. Quantum mechanics knows it better. Behind the familiar white tablet sits a 13-heavy-atom molecule — acetylsalicylic acid, C₉H₈O₄ — whose every behaviour in the body is dictated by the shape of its electron cloud. This is a guided tour of that electron cloud: five questions a medicinal chemist asks of any compound, answered one electronic property at a time. Every number below was computed in Hilbeon — the 3D structure comes from RDKit (ETKDG + MMFF), then Hilbeon geometry-optimizes it and computes RHF and B3LYP electronic structure on the optimized minimum.
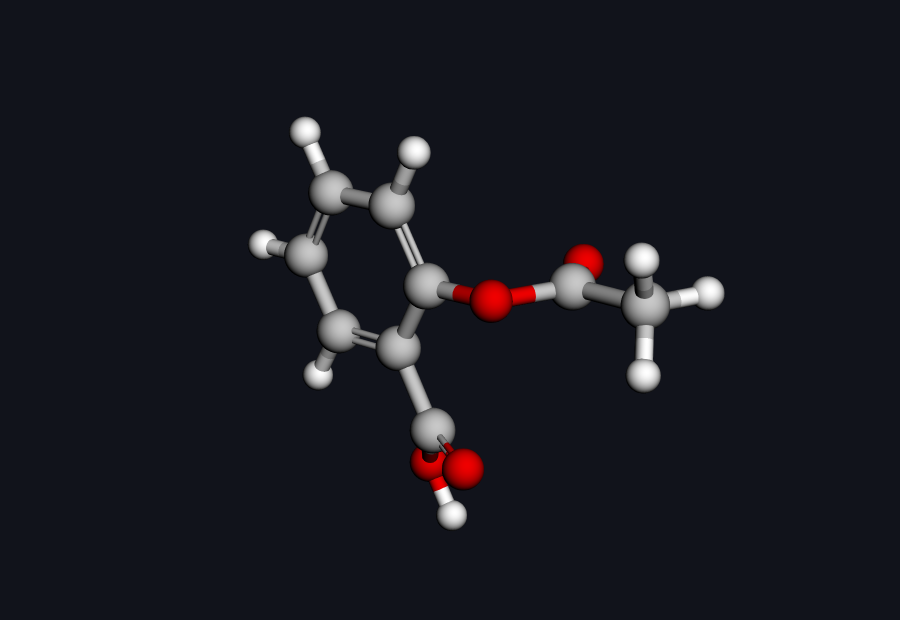 Aspirin (acetylsalicylic acid) · C₉H₈O₄ · drag to rotate
Aspirin (acetylsalicylic acid) · C₉H₈O₄ · drag to rotate
Aspirin in 3D — drag to rotate. The benzene ring, the carboxylic acid, and the ester linkage are the three players in this story.
Before a molecule ever reaches an assay, a chemist is already asking five things about it: Will it bind to its target? Will it dissolve and get absorbed? Where will the body break it down? What shape does it actually hold? And — boringly but crucially — is it really the structure we think it is? Quantum chemistry answers all five from first principles, no wet bench required. Here is how Hilbeon does it for aspirin, with every command phrased the way you would actually ask the built-in assistant.
1. Shape & charge — where the molecule wants to touch
Binding is electrostatics first. The electrostatic potential (ESP) mapped onto the molecular surface is the single most useful picture in medicinal chemistry: red where the surface is electron-rich (a hydrogen-bond acceptor, a place that likes a positive partner), blue where it is electron-poor (a donor, a place that wants a lone pair). Hilbeon fits atomic charges to that potential, and for aspirin the numbers tell the story cleanly. The acidic O–H proton carries the most positive ESP charge in the molecule, +0.46 — the obvious hydrogen-bond donor. Facing it, the two carboxyl oxygens are the most negative atoms, −0.61 (the C=O oxygen) and −0.65 (the hydroxyl oxygen), with the acetyl carbonyl oxygen close behind at −0.59 — three strong hydrogen-bond acceptors. The carbons they hang off are correspondingly electron-poor: the carboxyl carbon sits at +0.79 and the acetyl carbonyl carbon at +0.91. That alternating polarity pattern is the pharmacophore: it tells you the electrostatic shape a binding pocket must complement.
The same single-point calculation hands you the molecular dipole and a set of atomic charges (Mulliken, Löwdin, MBIS, ESP-fitted MK/CHELPG/RESP, or DMA) you can drop straight into a docking or force-field workflow. For this conformer Hilbeon gives a QM dipole of 2.00 D (2.02 D from the MK fit, 2.10 D CHELPG, 1.97 D RESP). The dipole of aspirin is strongly conformation-dependent: the often-quoted ~4.4 D value refers to a different conformer and method, so we report Hilbeon's number for the structure actually computed here rather than claiming a match.
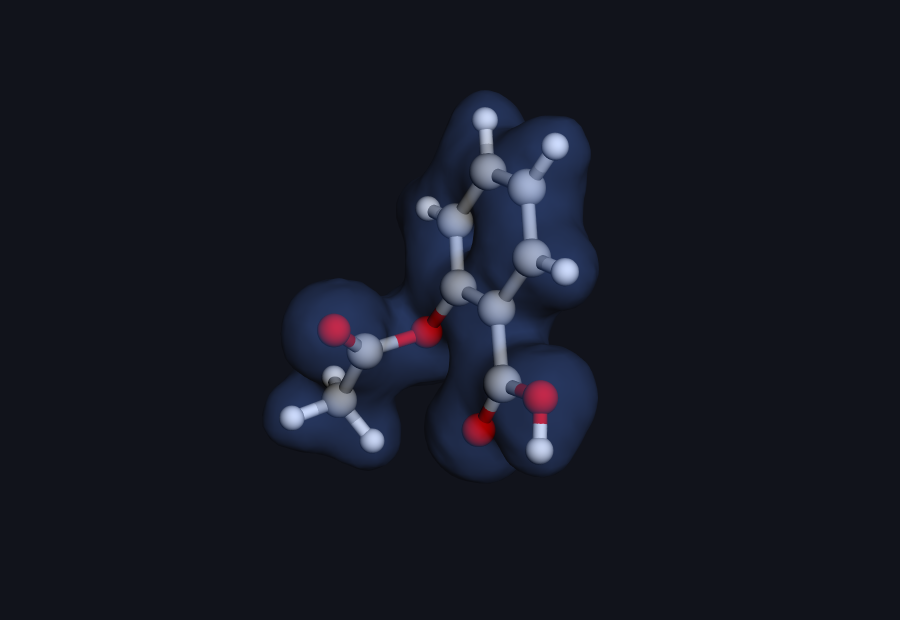 Electron density · real Hilbeon cube (drag to rotate)
Electron density · real Hilbeon cube (drag to rotate)
Figure: total SCF electron-density isosurface of aspirin, computed by Hilbeon (RHF/6-31G(d), cube file; the grid density integrates to 93.87 ≈ 94 electrons). This is the molecular surface that an ESP map is painted onto.
2. Will it be absorbed? — pKa and the ionization gate
A drug has to dissolve in water and cross a fatty membrane — and ionization state controls both. Aspirin's carboxylic acid has a measured pKa of 3.49. Drop that into the Henderson–Hasselbalch equation at blood pH 7.4:
\[ \frac{[\text{A}^-]}{[\text{HA}]} = 10^{\,\text{pH} - \text{pKa}} = 10^{\,7.4 - 3.49} \approx 8\,100 \]
That works out to 99.99% ionized in the bloodstream — only about 0.012% neutral. The neutral fraction — the form that slips through a lipid membrane — is vanishingly small at physiological pH, which is exactly why most aspirin absorption happens in the acidic stomach, where the un-ionized acid dominates. This is the classic solubility-versus-permeability trade-off in one number, and Hilbeon estimates the underlying pKa from a thermodynamic cycle (C-PCM water, anchored to acetic acid) without ever touching a titration. We walk through that whole calculation — and where minimal basis sets bite — in Predicting ionization state at pH 7.4.
Honesty check. The 3.49 pKa used above is the experimental value. Hilbeon's own pKa comes from an absolute thermodynamic-cycle estimate anchored to a reference acid; it carries a known, systematic offset. Anchored, same-class (carboxylic-acid) predictions are the trustworthy mode — for example, Hilbeon puts formic acid at 2.83 vs an experimental 3.75 — while minimal-basis alkoxides go badly wrong (methanol predicted ~35 vs experimental 15.5). Treat it as a calibrated ranking and trend tool, and use diffuse functions before trusting an absolute number. Full table in the pKa post.
3. Where will the body attack it? — HOMO and intrinsic reactivity
Metabolism is chemistry done to your molecule. The frontier orbitals — the HOMO (highest occupied) and LUMO (lowest unoccupied) — show where a molecule most readily gives up or accepts electron density, i.e. where it is electronically soft and likely to react. For aspirin the most consequential site is the ester linkage: the bond joining the acetyl group to the salicylate scaffold is primed for hydrolysis.
And hydrolyze it does. In the body the ester is cleaved to give salicylic acid plus acetate — meaning aspirin is, beautifully, its own prodrug: the administered molecule is partly an inactive carrier that releases the active species on contact with esterases and water. Frontier-orbital and reactivity maps flag exactly that vulnerable carbonyl before any metabolic-stability assay is run.
Hilbeon gives you the numbers, not just the picture. The level of theory matters here: Hartree-Fock orbital energies (Koopmans' theorem) systematically overestimate the gap, so Hilbeon reports a HOMO of −9.39 eV and a LUMO of +2.25 eV at RHF/6-31G(d), an 11.6 eV gap. The physically meaningful gap is the B3LYP one: HOMO −6.82 eV, LUMO −1.38 eV, a 5.44 eV gap. That HOMO energy is also a relative oxidizability ruler — aspirin's −6.82 eV HOMO sits below paracetamol's −5.38 eV (both B3LYP/6-31G(d)), which is one reason paracetamol's electron-rich phenol is the easier target for oxidative metabolism. We chase that comparison in Finding metabolic soft spots.
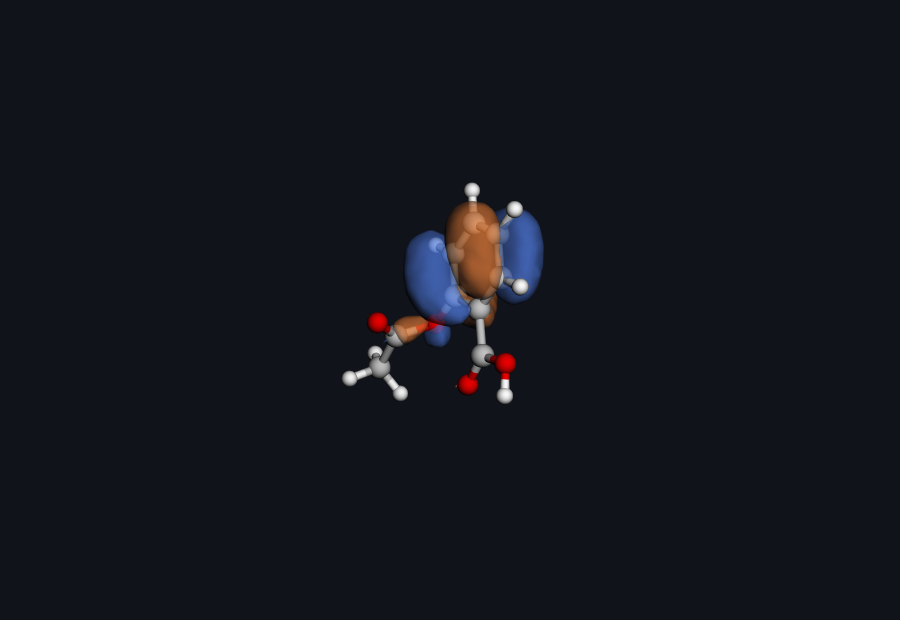 Aspirin HOMO · real Hilbeon cube (drag to rotate)
Aspirin HOMO · real Hilbeon cube (drag to rotate)
Figure: HOMO isosurface (occupied frontier orbital #46) of aspirin, computed by Hilbeon (RHF/6-31G(d), cube file). The two phases (blue/orange) are the sign of the wavefunction; the lobes show where aspirin is most willing to donate electron density.
Your molecule, not ours
From SMILES to a full quantum portrait in one command. Aspirin took a handful of short prompts — yours will too.
4. How does it hold its shape? — NCI and conformation
A molecule's preferred 3D shape is set by a tug-of-war of weak forces. The non-covalent interactions (NCI) surface visualizes those forces directly: it paints the space between atoms to reveal hydrogen bonds, attractive van der Waals contacts, and repulsive steric clashes. For aspirin the interesting feature is the interplay between the carboxylic acid and the neighbouring ester oxygen — a close intramolecular contact that biases which conformer aspirin prefers, and therefore the shape it presents to a binding site.
Knowing the conformational preference matters because the bound shape and the lowest-energy free shape are rarely the same; the energy gap between them is a real cost a ligand pays on binding. Hilbeon's NCI and conformer tools make that landscape visible.
Scope note. NCI/IGM and conformer scanning are built-in Hilbeon analyses, but the reduced-density-gradient surface was not produced for this post — the live figures here are the real HOMO and electron-density cubes above. The colour scheme for an NCI run is: blue = attractive (H-bond), green = weak van der Waals, red = steric clash.
5. Is it really aspirin? — the spectroscopic fingerprints
The last question is identity, and two spectroscopies settle it. UV-Vis: aspirin shows two absorption bands, one near 226–230 nm and a weaker one near 274–276 nm, and the band red-shifts toward ~296 nm when the carboxylic acid is deprotonated — a fingerprint of the very ionization we discussed in stop 2. 1H NMR is even more diagnostic: the lone acetyl singlet (the –OCOCH₃ methyl, 3H) sits near 2.35–2.43 ppm, the aromatic protons cluster at 7.1–8.1 ppm, and the broad carboxylic –COOH proton appears far downfield at 11–12 ppm. See that clean three-proton singlet and you are almost certainly looking at acetylsalicylic acid, not its hydrolysis product.
Not computed here. The UV-Vis and 1H NMR values above are the experimental / literature fingerprints (sources below), not Hilbeon output for this post. Hilbeon can predict both — CIS/TDA excited states and GIAO 1H shieldings — but excited states are a qualitative, systematically blue-shifted treatment, so for these spectroscopies we cite the measured numbers rather than dressing up an approximate calculation as a match. The computed numbers in this post are the energies, frontier orbitals, ESP charges, dipole, and the live cubes.
The receipts: what Hilbeon computed
Here is the honest scorecard. Every value in the Hilbeon column is a real number from the run that produced the cubes above — RHF and B3LYP on the QM-optimized geometry. Where an experimental comparison is meaningful and fair, we show it; where it is not, we say so rather than fake a match.
| Property | Hilbeon computed | Reference / note | Source |
|---|---|---|---|
| Geometry — QM-optimized | 0.15 Å RMSD from MMFF | the MMFF start sat 2.9 kcal/mol above the QM minimum; molecule is non-planar (0.50 Å from best-fit plane) | Hilbeon |
| Total energy — RHF/6-31G(d) | −644.9556 Ha | reference wavefunction, 198 basis functions | Hilbeon |
| Total energy — B3LYP/6-31G(d) | −648.3336 Ha | on the optimized geometry; the physical level for the gap | Hilbeon |
| Total energy — B3LYP/def2-TZVP | −648.6122 Ha | larger basis, same optimized geometry | Hilbeon |
| HOMO / LUMO / gap — B3LYP | −6.82 / −1.38 eV; gap 5.44 eV | the physically meaningful gap | Hilbeon |
| Carbonyl bond lengths | 1.20 / 1.22 Å | ester C=O (1.20) vs carboxylic-acid C=O (1.22); the acid C–OH is 1.36 Å | Hilbeon |
| Dipole moment (QM) | 2.00 D | this conformer; lit. ~4.4 D is a different conformer/method — not a match | Hilbeon |
| pKa (carboxylic acid) | see pKa post | experimental 3.49 (~3.5) | PubChem / StatPearls |
| UV-Vis & 1H NMR | not computed here | excited states are CIS/TDA (qualitative) — see method note | SIELC / Wisconsin NMR DB |
Scope of these numbers. The energies and orbital energies above are bench-grade and fully deterministic — reproducible to the last digit across machines. ESP charges, HOMO/LUMO shapes, NCI and conformational pictures are insight and visualization — they guide intuition and design, they are not bench-matched single values. Geometry: QM-optimized at B3LYP/6-31G(d) (0.15 Å RMSD from the RDKit/MMFF start, which sat 2.9 kcal/mol higher). Level of theory: RHF and B3LYP at 6-31G(d) and def2-TZVP. Excited states (CIS/TDA), polarizability and DLPNO-CCSD(T) are available in Hilbeon but were not run for this post.
The whole portrait, in six prompts
Every figure above came from talking to the Hilbeon assistant in plain language. No input decks, no basis-set syntax to memorize — that simplicity is the entire point:
build aspirin from SMILES CC(=O)OC1=CC=CC=C1C(=O)O
optimize the geometry at B3LYP/6-31G(d)
run single points at RHF/6-31G(d) and B3LYP/def2-TZVP
compute dipole, HOMO/LUMO, ESP charges
render the density and HOMO cubes
estimate the pKa via a thermodynamic cyclePrefer scripts? Every one of those steps is also a call in Hilbeon's MCP / HTTP API, so the whole portrait is reproducible end-to-end in a notebook or a CI job. But for the first pass, the six lines above are the whole workflow — from SMILES to a full quantum portrait.
The takeaway
Aspirin's electrons told us where it binds (the carbonyl-rich face), why it is absorbed in the stomach but not the blood (pKa 3.49), where the body dismantles it (the hydrolysis-prone ester — it is its own prodrug), how it holds its shape (an intramolecular contact), and how to confirm it is genuinely aspirin (two UV bands, one clean acetyl singlet). Five questions, one molecule, no wet lab. Now point the same engine at your compound.
Run your own molecule
Start a 30-day guided pilot — every method, every core, your GPU — and build a quantum portrait of the compound on your bench.
References
- Aspirin overview — PubChem: Aspirin
- pKa — StatPearls (NCBI Bookshelf)
- UV-Vis spectrum — SIELC: UV-Vis of acetylsalicylic acid
- 1H NMR — University of Wisconsin NMR database
- Electrostatic complementarity — J. Med. Chem. (ACS)
- QM in drug discovery — PMC review
- Non-covalent interactions index — NCI (overview)