Application note
Predicting Substituent Effects: a Hammett Plot from QM
Substituent effects Reactivity Drug design
Almost every molecule a medicinal chemist makes is a variation on the last one — swap a methyl for a chlorine, a methoxy for a nitrile, push and pull electrons around the same scaffold. The question that decides which analogue to make next is always the same: how will this substituent change the molecule?
Will it make the compound more acidic? More electrophilic? Bind tighter, or get chewed up faster by a P450? Remarkably, a single number — the Hammett constant σ — predicts a startling amount of all of this, and it has done so since the 1930s. This post shows how Hilbeon regenerates that ninety-year-old empirical scale from first principles, straight out of the electron density, and why that lets you estimate the pull of a substituent that has no tabulated σ at all.
What the Hammett constant actually is
In 1937 Louis Hammett noticed that if you take benzoic acid and hang a substituent on the para position, the change in its acidity is remarkably transferable: the same substituent shifts the rate or equilibrium of a completely different reaction by a proportional amount. He distilled that into a constant σ for each group, defined empirically from the aqueous pKa of substituted benzoic acids. Electron-withdrawing groups got a positive σ; electron-donating groups a negative one. Ninety years later, σ is still one of the first things a chemist reaches for when designing an analogue series.
The physical picture is simple. Deprotonate a benzoic acid and you make a negatively charged carboxylate. Anything that helps spread out or stabilise that extra negative charge makes the acid stronger. An electron-withdrawing group para to the carboxyl pulls density toward itself, props up the anion, and lowers the energy cost of letting the proton go. An electron-donating group does the opposite. σ is, at bottom, a measure of that electronic push or pull — and electronic structure is exactly what Hilbeon computes.
Reproducing the scale from the wavefunction
We took the canonical Hammett series — seven para-substituted benzoic acids spanning strong donors to strong acceptors — and, for each one, computed the gas-phase deprotonation energy ΔE: the energy difference between the neutral acid and its carboxylate anion. That carboxylate is the hard part. A bare anion has diffuse, loosely-held electron density that a standard basis set simply cannot describe, so the calculation uses diffuse-augmented basis functions (aug-cc-pVDZ) built to reach out into that tail. Then we plotted ΔE against each group's tabulated σ.
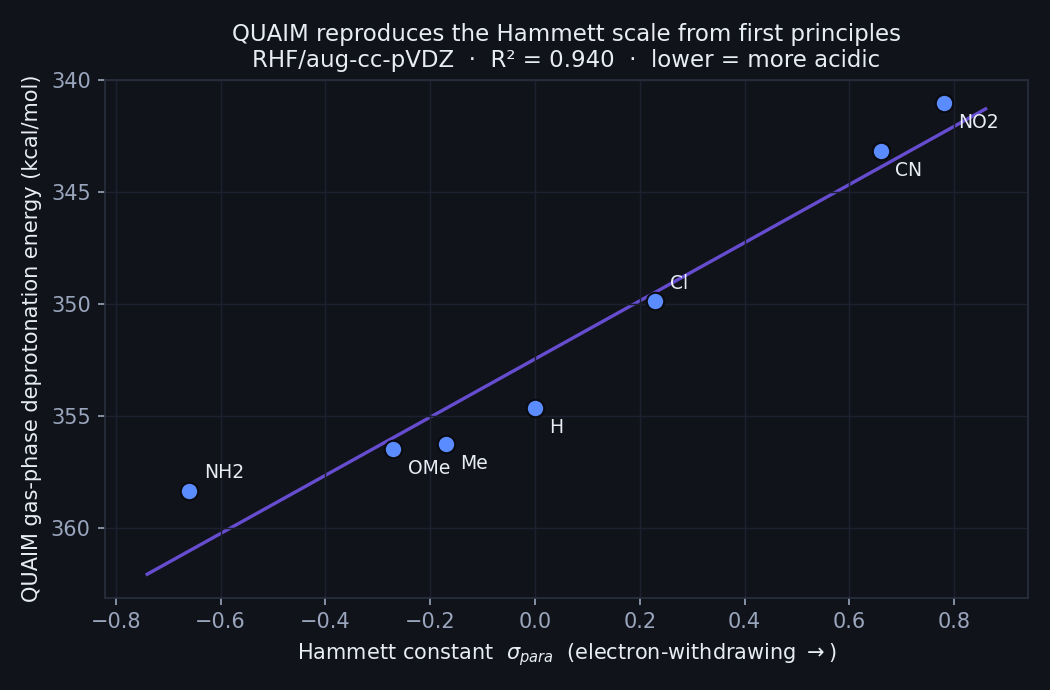
Hilbeon's gas-phase deprotonation energy vs the Hammett σ (RHF/aug-cc-pVDZ). A clean linear trend (R² = 0.94): electron-withdrawing groups (right) make the acid stronger.
The trend is about as clean as physical-organic chemistry ever gets. As σ climbs from −0.66 (the strong donor –NH2) to +0.78 (the strong acceptor –NO2), the deprotonation energy falls almost in a straight line — a fitted slope of −13.0 kcal/mol per unit σ, with R² = 0.94. Every electron-withdrawing substituent makes the acid stronger; every electron-donating one makes it weaker. Hilbeon never saw a single experimental pKa: the ordering falls straight out of the computed electron density.
| Substituent (para) | σpara | Hilbeon ΔEdeprot (kcal/mol) |
|---|---|---|
| –NH2 | −0.66 | 358.3 |
| –OMe | −0.27 | 356.5 |
| –Me | −0.17 | 356.2 |
| –H | 0.00 | 354.7 |
| –Cl | 0.23 | 349.9 |
| –CN | 0.66 | 343.2 |
| –NO2 | 0.78 | 341.1 |
Read top to bottom, the deprotonation energy spans 17.3 kcal/mol from the most electron-rich acid to the most electron-poor — the entire electronic effect of the substituent, laid out on one axis. The donors (–NH2, –OMe, –Me) cluster at the high-energy, harder-to-deprotonate end; the acceptors (–Cl, –CN, –NO2) at the low-energy, easier-to-deprotonate end, in the same order a Hammett table would give you. Unsubstituted benzoic acid (–H, σ = 0) sits, by construction, right in the middle.
Score your own substituents
From a list of SMILES to a Hammett-style plot in a couple of prompts. Read the electronic pull of your groups — even ones with no tabulated σ — before you commit to a synthesis.
Why this is more than a re-derivation
Reproducing a known scale is a nice validation, but the real payoff is what it unlocks. The Hammett table is finite — it covers the few hundred substituents someone has bothered to measure. The moment you design a group nobody has tabulated — a fused heterocycle, an unusual fluorinated motif, a fragment from a generative model — the table is silent. The Hilbeon calculation is not. Because it reads the electronic effect directly from the density, you can place a brand-new substituent on the same axis and ask where it lands: is it pulling like a nitrile, or pushing like a methoxy?
That is the deeper reason the gas-phase numbers track an aqueous-derived scale so tightly. The substituent effect captured here is largely intrinsic and electronic — a property of how the group reshapes the molecule's charge distribution — which is precisely why σ transfers from one reaction to another, and from solution to gas phase, in the first place. The same electronic pull that shifts a pKa also nudges binding, conjugation, and metabolic lability, which is why σ correlates with so much across medicinal chemistry.
What this is — and is not. These are RHF/aug-cc-pVDZ single-point energies on RDKit MMFF geometries — the structures were generated by force field and given a Hilbeon single-point electronic-structure calculation; they were not QM-reoptimized. ΔE is a gas-phase deprotonation energy, while Hammett σ is defined from aqueous pKa. The strong linear correlation shows the substituent effect is largely intrinsic and electronic (which is exactly why it transfers from solution to gas phase) — but the fitted slope of −13.0 kcal/mol per σ is not the aqueous reaction constant ρ, and a gas-phase ΔE is not an aqueous pKa. To turn this into absolute solution acidity, refine with DFT or MP2, optimize the geometries, and add a solvation model (C-PCM / SMD) with thermal corrections.
Why diffuse functions matter here. Every point on this plot involves a carboxylate anion, and anions carry diffuse, loosely-bound electron density that a standard basis set underdescribes — get it wrong and the whole acidity trend tilts. Hilbeon runs these on its own integral engine (McMurchie–Davidson + Rys — no third-party integral libraries) and is numerically validated to tight tolerances and bit-identical across machines on exactly these hard anionic cases to roughly 10−9 Ha: phenyltetrazolate, for instance, comes in at −485.8211471002, fully deterministic and reproducible to the last digit across machines. The diffuse-augmented anions you are ranking are the real ones.
How to do it yourself
Every number above came from talking to the Hilbeon assistant in plain language — no input decks, no basis-set syntax to memorize:
build 4-nitrobenzoic acid from SMILES
deprotonate it to the carboxylate
run RHF/aug-cc-pVDZ on both the acid and the anion
report the deprotonation energy in kcal/mol
repeat for the NH2, OMe, Me, H, Cl and CN analogues
plot deprotonation energy against the Hammett sigma and fit a linePrefer scripts? Every one of those steps is also a call in Hilbeon's MCP / HTTP API, so screening a whole library of substituents is a loop in a notebook or a CI job. Point it at a new motif with no tabulated σ and read its electronic pull straight off the same axis.
The takeaway
The Hammett constant is one of the oldest and most useful predictive tools in chemistry, and for ninety years it has been an empirical one — a lookup table built from aqueous pKa measurements. Hilbeon regenerates the same ordering from nothing but the electron density: seven para-substituted benzoic acids, a gas-phase deprotonation energy each, and a clean linear correlation against σ at R² = 0.94, with electron-withdrawing groups stabilising the anion and electron-donating groups doing the opposite. Treat it as an intrinsic-electronic-effect tool, not an absolute-pKa oracle — refine with DFT/MP2, geometry optimization and solvation when you need real solution numbers — and you have a way to estimate the pull of a substituent before anyone has measured it. Point the same engine at your own analogue series and read the substituent effect off the density.
Score your own substituents
Start a 30-day guided pilot — every method, every core, your GPU — and read the electronic effect of the groups on your scaffold before you make them.
References
- The original Hammett correlation — L. P. Hammett, J. Am. Chem. Soc. 1937, 59, 96; and Physical Organic Chemistry (McGraw-Hill, 1940).
- Tabulated σ constants — C. Hansch, A. Leo & R. W. Taft, “A Survey of Hammett Substituent Constants and Resonance and Field Parameters,” Chem. Rev. 1991, 91, 165.
- Substituent constants in QSAR and drug design — Hammett equation (overview).
- Spreading the anion the other way — Carboxylic Acid vs Tetrazole: a Bioisostere Up Close.