Application note
Carboxylic Acid vs Tetrazole: Reading a Classic Bioisostere
Bioisosteres Permeability Drug design
A bioisostere swap is the medicinal chemist's polite con: replace one functional group with another that keeps the property that matters — the binding interaction, the acidity — while quietly fixing something that doesn't, like membrane permeability or metabolic liability. The textbook example is swapping a carboxylic acid (−COOH) for a 1H-tetrazole. This post shows why that swap works, read straight off the electron density by Hilbeon.
The premise is that the two groups are nearly interchangeable where it counts. Benzoic acid has an aqueous pKa of about 4.2; 5-phenyltetrazole comes in around 4.5. At physiological pH both are essentially fully ionized, so the anion that docks into a target's positively charged pocket is preserved — same charge, same salt bridge, same recognition. Yet the tetrazole version is frequently more permeable and more metabolically stable. That is not folklore: the blockbuster “sartan” antihypertensives — losartan, valsartan, candesartan, irbesartan — all carry a tetrazole exactly where a carboxylic acid would otherwise sit. The question is the interesting one: if the acidity is matched, where does the permeability advantage come from?
The answer lives in the anion
Whatever the two neutral acids do, the species that has to cross a membrane — and pay the price for being charged — is the deprotonated anion. To cross a lipid bilayer, an ion must shed part of its hydration shell. The energetic cost of that stripping, the desolvation penalty, scales with how tightly the molecule grips its water, which in turn scales with local charge density: a −1 charge crammed onto a couple of atoms holds water far more fiercely than the same −1 smeared thinly over many atoms.
So the right comparison is not the two acids but the two conjugate bases — benzoate and 5-phenyltetrazolate — and the right question is purely electronic: where does the −1 charge actually go? That is a question about the wavefunction, and it is exactly what Hilbeon was built to answer. Anions, though, are notoriously hard: their loosely bound electron cloud puffs out far from the nuclei, and a standard basis set simply has no functions out there to describe it. You need diffuse-augmented basis sets — the aug-cc-pVDZ family and friends — or the anion's energy and charge distribution are both wrong. Hilbeon now ships those bases, so we can put the two anions on an honest footing.
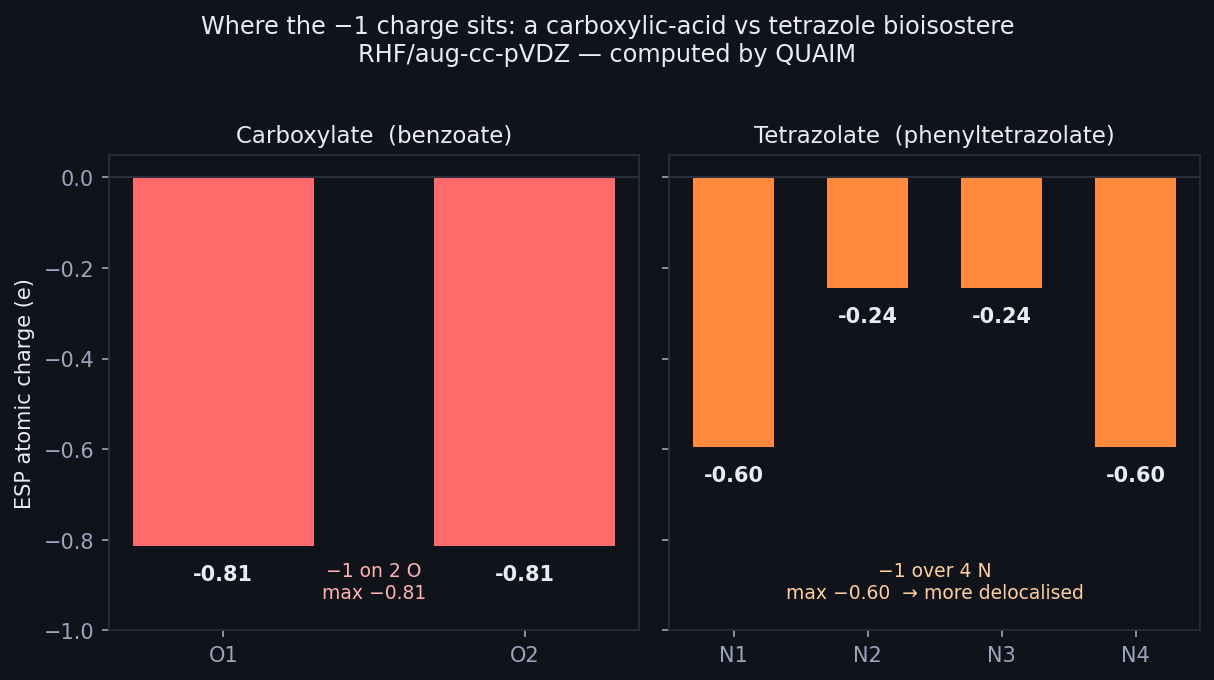
Where the −1 charge sits (RHF/aug-cc-pVDZ, Hilbeon). Carboxylate: −0.81 on each of 2 O. Tetrazolate: spread over 4 N (max −0.60) — more delocalised, easier to desolvate.
The reveal: two oxygens vs four nitrogens
Here is the whole story in one sentence. The carboxylate has nowhere to hide its charge: the −1 piles onto just two oxygens, each carrying an ESP charge of roughly −0.81. The tetrazolate, by contrast, is a five-membered ring with four nitrogens, and the −1 spreads over all four — the two most negative N reach only −0.60, and the remaining two sit near −0.24 and −0.25. Same total charge, very different geometry of charge.
That single difference — peak local charge of 0.81 on two atoms versus 0.60 spread over four — is the bioisostere's secret. Lower local charge density means a weaker grip on the surrounding water, which means a smaller desolvation penalty to slip into a membrane, which means better passive permeability — and all of it at matched aqueous acidity, so the binding interaction the chemist worked so hard for is untouched. The tetrazole isn't a better acid; it's a more delocalised one, and delocalisation is what crosses membranes.
 Benzoate anion — drag to rotate
Benzoate anion — drag to rotateBenzoate (C7H5O2−). The −1 has only two oxygen atoms to live on — high local charge density, a tight hydration shell.
 5-Phenyltetrazolate anion — drag to rotate
5-Phenyltetrazolate anion — drag to rotate5-Phenyltetrazolate (C7H5N4−). Four ring nitrogens share the −1 — the charge is smeared out, the water held more loosely.
Compare your own bioisosteres
From two SMILES strings to two anion charge maps in a couple of prompts. See where the charge actually goes before you commit a synthesis.
The receipts
Three numbers carry the argument: the acidities are matched in water, the gas-phase trend exposes the delocalisation, and the charge maps show where it comes from.
| Observable | Carboxylic acid / benzoate | Tetrazole / tetrazolate |
|---|---|---|
| Aqueous pKa (experiment) | 4.20 | ~4.5 |
| Hilbeon gas-phase deprotonation energy (kcal/mol) | 354.7 | 342.7 |
| Charge delocalisation: max |q| (atoms sharing −1) | 0.81 (2 O) | 0.60 (4 N) |
Read the table top to bottom. In water the two pKa values are essentially equal — that is the bioisostere premise, and it is why the swap preserves binding. In the gas phase, where there is no solvent to even the score, the tetrazole is the intrinsically stronger acid by 12.0 kcal/mol, precisely because its anion is stabilised by spreading the charge over four nitrogens. The two facts are the same fact seen twice: the very delocalisation that makes tetrazolate easier to form in the gas phase is the delocalisation that makes it easier to desolvate in a membrane. The charge-delocalisation row is the mechanism made visible.
Trust the anion. Diffuse-heavy anions are the acid test of a quantum-chemistry engine, so we checked. On the 5-phenyltetrazolate anion, Hilbeon's owned integral engine is fully deterministic and reproducible to the last digit across machines — −485.8211471002 Ha at aug-cc-pVDZ, stable to nine figures. The hard case is numerically rock-solid, so the charge map you are reading is the real one.
What these numbers are — and are not. Everything here is RHF/aug-cc-pVDZ, a single point on RDKit/MMFF geometries — the structures were generated and force-field-relaxed by RDKit and given a Hilbeon electronic-structure calculation; they were not QM-reoptimised. The gas-phase deprotonation energy is an intrinsic / relative acidity trend: tetrazole is the stronger acid in the gas phase, while in water the two are roughly equal because solvation differs between them. It is not an absolute aqueous pKa — that needs an explicit solvation model (SMD or C-PCM) plus thermal corrections, a natural refine target. Finally, we deliberately do not report a “dipole” for either anion: for a charged species the dipole depends on where you put the coordinate origin, so it is physically meaningless. The robust, qualitative observable is the charge delocalisation, and that is what the figures show.
How to do it yourself
Every number above came from talking to the Hilbeon assistant in plain language — no input decks, no basis-set syntax to memorize:
build benzoic acid from SMILES OC(=O)c1ccccc1, then deprotonate it
run RHF/aug-cc-pVDZ on the benzoate anion
report the ESP atomic charges and render the charge map
repeat for 5-phenyltetrazole -> 5-phenyltetrazolate
compare the gas-phase deprotonation energiesPrefer scripts? Every one of those steps is also a call in Hilbeon's MCP / HTTP API, so screening a whole set of acid bioisosteres is a loop in a notebook or a CI job. Hilbeon runs on its own GPU-accelerated integral engine (McMurchie–Davidson + Rys — no third-party integral libraries), numerically validated to tight tolerances of roughly 1e−9 Ha and bit-identical across machines, even for the diffuse-heavy anions that break lesser setups, so the charges you are reading are the real ones.
The takeaway
A bioisostere is a trade, and the classic carboxylic-acid–to–tetrazole swap is a good one because it pays in permeability without spending acidity. Hilbeon shows you the ledger: matched aqueous pKa, a gas-phase trend that exposes the tetrazolate's extra stability, and a charge map that explains both at once — −0.81 on two oxygens versus a gentle −0.60 across four nitrogens. Lower local charge density, smaller desolvation penalty, easier passage through a membrane, same binding charge at the target. That is the sartans' design choice, and it is visible in nothing more than the electron density. Point the same engine at your own acidic warts and see which bioisostere actually moves the charge before you make a single analogue.
Compare your own bioisosteres
Start a 30-day guided pilot — every method, every core, your GPU, diffuse bases for anions — and read the charge maps of the groups on your bench before you commit a synthesis.
References
- Bioisosterism, a review — N. A. Meanwell, J. Med. Chem. 2011, 54, 2529 (and the 2018 update, J. Med. Chem. 2018, 61, 5822)
- Tetrazole as a carboxylic-acid bioisostere — C. G. Wermuth review of acid surrogates; tetrazole (acidity & bioisosterism)
- The sartans — losartan, valsartan, candesartan, irbesartan (all carry a tetrazole)
- Desolvation and passive permeability — the lipophilicity / charge-density rationale for ion transport across membranes
- The acid side of the same coin — What ionization state is your molecule at pH 7.4?