Application note
Electron-Rich or Electron-Poor? The Rings in Your Drugs
Heterocycles Ring design ADMET
Choosing the aromatic ring at the heart of a molecule is one of the most consequential decisions in a drug's design. That one ring quietly sets how the compound stacks, where it gets oxidised, and how it hydrogen-bonds — and almost all of it is governed by a single property you can compute: how electron-rich the ring is.
Swap a phenyl for a pyridine and you have not just moved an atom. You have lowered the ring's electron density, turned a π-donor into a π-acceptor, added an H-bond acceptor and a basic nitrogen, and made the ring markedly harder for a cytochrome P450 to oxidise. Medicinal chemists carry an intuition for this from experience. This post shows how Hilbeon puts ten of the most common drug rings onto a single frontier-orbital ladder, turning that intuition into a number you can rank.
The ring's electron-richness lives in its frontier orbitals
An aromatic ring's chemistry is dominated by its two frontier orbitals: the highest occupied (HOMO) and the lowest unoccupied (LUMO). The HOMO is the cloud of electrons the ring offers up to the world — the higher (less negative) its energy, the more loosely those electrons are held, and the more electron-rich the ring. That single fact branches into the three properties a medicinal chemist cares about most:
- π-stacking. An electron-rich ring (high HOMO) is a good π-donor; an electron-poor ring (low HOMO and LUMO) is a π-acceptor. The strongest aromatic stacking pairs a donor with an acceptor, so knowing which side of the ladder a ring sits on tells you how to strengthen a stacking interaction in a binding pocket.
- Metabolic-oxidation liability. Cytochrome P450 enzymes attack electron-rich aromatics — a high HOMO is exactly the soft target their oxidative chemistry seeks out. Electron-rich rings are oxidised faster; electron-poor rings are more metabolically robust.
- H-bonding and basicity. The same nitrogens that pull a ring's HOMO down also add hydrogen-bond acceptors and tune basicity, reshaping solubility and the molecule's interaction map.
So a ring with a high HOMO is an electron-rich, generous π-donor — great for stacking, but a soft spot for oxidative metabolism. A ring with a low HOMO and LUMO is electron-poor: a π-acceptor, and metabolically tougher. Hilbeon reads both numbers straight off the wavefunction.
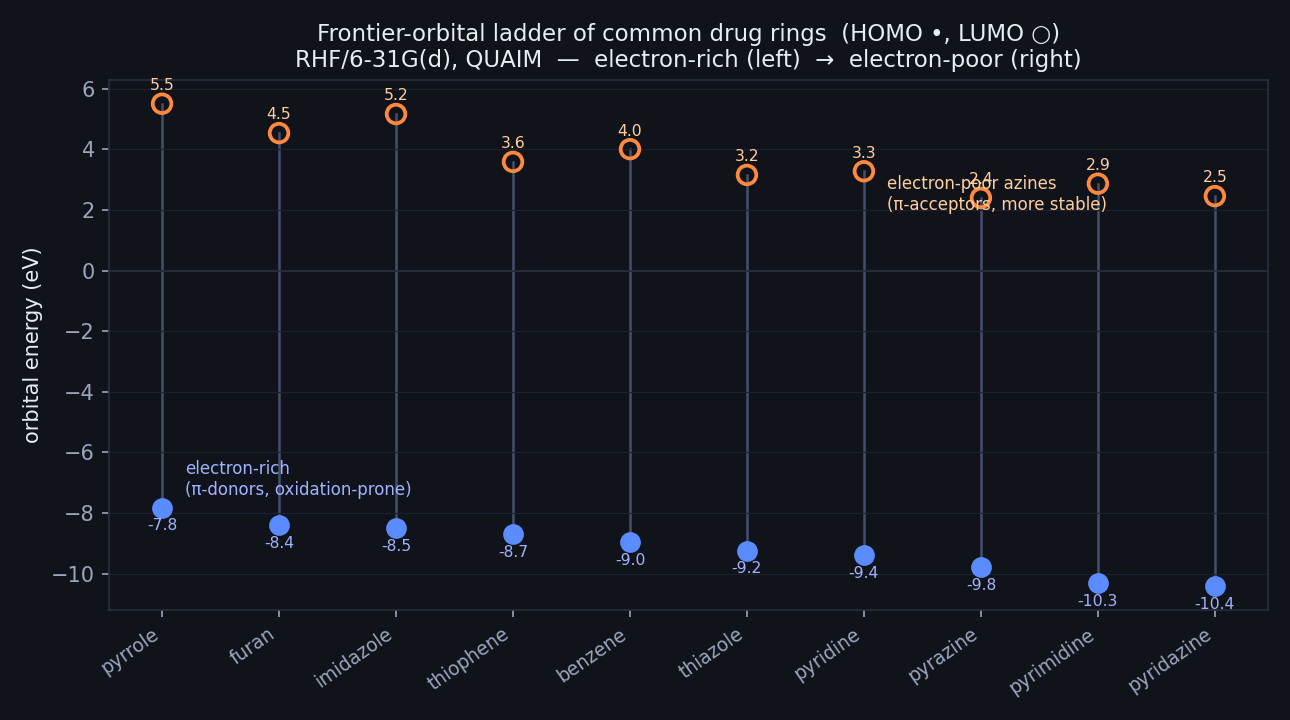
Frontier-orbital ladder of ten common drug rings (RHF/6-31G(d), Hilbeon). HOMO (filled) and LUMO (open), ordered electron-rich (left) → electron-poor (right).
Ten rings on one ladder
We took ten of the workhorse aromatic rings of medicinal chemistry — the five-membered heterocycles, the diazines, pyridine, and benzene as the reference — and computed the frontier orbitals of each. Ordered by HOMO energy, they fall into a clean, chemically sensible sequence.
At the electron-rich top sit the five-membered π-excessive rings: pyrrole leads at a HOMO of −7.83 eV, followed by furan (−8.40), imidazole (−8.51) and thiophene (−8.70). These rings push six π-electrons through five atoms, so the density per atom is high and the HOMO rides up — excellent π-donors, and the usual suspects for oxidative metabolism.
At the electron-poor bottom sit the six-membered azines, where each ring nitrogen pulls density out of the π-system: pyridine (−9.40), then the diazines — pyrazine (−9.80), pyrimidine (−10.32) and pyridazine (−10.41). Every added nitrogen drops the HOMO further. These are the π-acceptors, and the metabolically robust end of the scale. Benzene (−8.97) and thiazole (−9.25) sit in between, bridging the π-excessive heterocycles and the electron-poor azines.
| Ring | HOMO (eV) | LUMO (eV) | Gap (eV) |
|---|---|---|---|
| Pyrrole | −7.83 | 5.51 | 13.34 |
| Furan | −8.40 | 4.54 | 12.94 |
| Imidazole | −8.51 | 5.17 | 13.68 |
| Thiophene | −8.70 | 3.59 | 12.29 |
| Benzene | −8.97 | 4.01 | 12.98 |
| Thiazole | −9.25 | 3.16 | 12.41 |
| Pyridine | −9.40 | 3.28 | 12.68 |
| Pyrazine | −9.80 | 2.40 | 12.20 |
| Pyrimidine | −10.32 | 2.87 | 13.19 |
| Pyridazine | −10.41 | 2.46 | 12.87 |
Read top to bottom, the HOMO spans 2.58 eV from the most electron-rich ring (pyrrole) to the most electron-poor (pyridazine) — a wide, smoothly graded axis to design along. The azines also push the LUMO down relative to benzene's 4.01 eV (pyrazine reaches 2.40): adding ring nitrogens makes the ring both a worse electron donor and, broadly, a better electron acceptor — though the exact LUMO ordering among the diazines is not strictly monotonic (the HF virtual is only qualitative).
Rank your own scaffolds
From a list of SMILES to a frontier-orbital ladder in a couple of prompts. Place your fused and exotic rings on the same axis — donor or acceptor, soft spot or stable — before you commit to a scaffold.
A ring-selection map you can act on
The point of putting every ring on one axis is that the axis becomes a design tool. Two moves fall straight out of it:
- Block a metabolic soft spot. If a series is being chewed up by P450 oxidation at an electron-rich ring — a pyrrole or thiophene high on the ladder — swap it for an electron-poor azine lower down. Moving from pyrrole (−7.83 eV) to pyrimidine (−10.32) pulls the HOMO down by two and a half electron-volts, starving the oxidative chemistry of the electron density it feeds on.
- Strengthen a stacking interaction. If the ring is meant to stack against an electron-rich aromatic in the pocket (a Trp or Tyr face), reach for a π-acceptor at the bottom of the ladder — a diazine — to deepen the donor–acceptor complement. If you need the ring itself to be the donor, stay up among the five-membered heterocycles.
These trade-offs are usually in tension — the electron-rich ring that stacks beautifully is the one P450 loves — and that is exactly why having both numbers in front of you matters. The ladder does not make the decision for you, but it shows you the whole board at once, with your own candidate rings placed on it.
What these numbers are — and are not. These are RHF/Koopmans frontier-orbital energies from RHF/6-31G(d) single points on RDKit MMFF geometries — the rings were built by force field and given a Hilbeon single-point electronic-structure calculation; they were not QM-reoptimized. They are excellent for trends and ranking — which ring is more electron-rich, which way a swap moves you — but they are not absolute ionisation potentials or electron affinities. The Hartree–Fock virtual orbital that gives the LUMO is especially qualitative; treat the LUMO column as directional, not quantitative. For quantitative orbital energies, refine with DFT (or outer-valence GW).
Why the rankings are trustworthy. Frontier-orbital comparisons only mean something if the underlying energies are clean, and Hilbeon computes these on its own integral engine (McMurchie–Davidson + Rys — no third-party integral libraries), fully deterministic and reproducible to the last digit across machines. The ladder is a like-for-like comparison — same method, same basis, same geometry protocol for all ten rings — so the ordering is solid even where the absolute orbital energy is only qualitative.
How to do it yourself
Every number above came from talking to the Hilbeon assistant in plain language — no input decks, no basis-set syntax to memorize:
build pyrrole, furan, imidazole, thiophene, benzene,
thiazole, pyridine, pyrazine, pyrimidine and pyridazine from SMILES
run RHF/6-31G(d) on each ring
report the HOMO and LUMO energies in eV
sort the rings from highest HOMO to lowest
plot them as a frontier-orbital ladderPrefer scripts? Every one of those steps is also a call in Hilbeon's MCP / HTTP API, so placing a whole library of candidate scaffolds on the ladder is a loop in a notebook or a CI job. Point it at a fused or exotic ring with no textbook intuition and read its electron-richness straight off the same axis as the rings you already know.
The takeaway
The aromatic ring you build a molecule around is a single decision that ripples through stacking, metabolism, and H-bonding — and all three are downstream of one computable property, how electron-rich the ring is. Hilbeon reads that property straight off the frontier orbitals and lines ten common drug rings up on one ladder: the five-membered π-excessive rings (pyrrole, furan, imidazole, thiophene) at the electron-rich, π-donor, oxidation-prone top; the six-membered azines (pyridine and the diazines) at the electron-poor, π-acceptor, metabolically robust bottom; benzene and thiazole in between. Treat it as a ranking tool, not an absolute-IP oracle — refine with DFT or GW when you need quantitative orbital energies — and you have a ring-selection map: swap an oxidation-prone ring for an azine to block a soft spot, or pick a π-acceptor to deepen a stacking interaction. Point the same engine at your own scaffolds and read the electronics off the density.
Rank your own scaffolds
Start a 30-day guided pilot — every method, every core, your GPU — and place the rings on your scaffolds on the same frontier-orbital ladder before you make them.
References
- N-heterocycles in FDA-approved drugs — E. Vitaku, D. T. Smith & J. T. Njardarson, “Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals,” J. Med. Chem. 2014, 57, 10257.
- Cytochrome P450 oxidation of electron-rich aromatics — P. R. Ortiz de Montellano, “Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes,” Chem. Rev. 2010, 110, 932.
- Aromatic π-stacking and the role of ring electronics — Aromatic stacking interactions (overview).
- Pulling the HOMO down the other way — Where Will This Molecule Get Metabolised? Mapping the Soft Spots.