Methods that matter
The Conformer Your Force Field Got Wrong
Conformers Dispersion DFT
Your conformer ranking is only as good as the weakest force in your energy model — and the weakest force is usually the one holding the molecule folded. London dispersion — the faint, universal attraction between any two close atoms — is what lets a flexible molecule tuck an aromatic ring against another group and gain energy. Force fields treat it with a crude pairwise term; plain, uncorrected DFT functionals like B3LYP miss it almost entirely. Rank conformers with either and you can confidently promote the wrong one to “most stable.”
Here is that failure in miniature, with every number computed.
One molecule, two shapes
Take bibenzyl — 1,2-diphenylethane, two phenyl rings on a short ethylene tether. It is the minimal model of the diaryl motif that runs through kinase inhibitors, antihistamines and countless other drugs. It has two archetypal conformers: an extended (anti) form with the rings apart, and a folded (gauche) form where the rings swing face-to-face, ~5 Å apart, close enough to feel each other's dispersion. Which one is lower in energy? We asked three models the same question, on the identical pair of geometries.
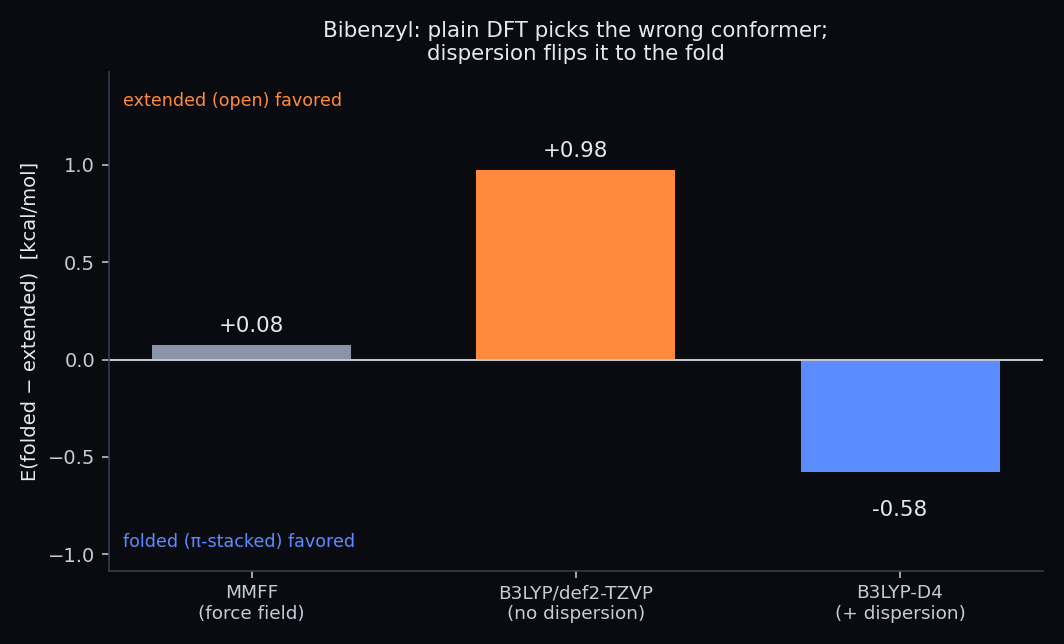
Relative conformer energy E(folded−extended) for bibenzyl. Positive = the open/extended form is favored; negative = the π-stacked fold is favored. The force field calls it a tie, plain DFT confidently picks the extended form, and adding dispersion (D4) flips the answer to the fold.
Reading the ladder
Three verdicts, three different answers:
- MMFF (a force field) puts the two within 0.08 kcal/mol — effectively a coin-flip. It can't resolve which conformer is real.
- B3LYP/def2-TZVP with no dispersion correction confidently favors the extended conformer by 0.98 kcal/mol. A clean, converged quantum calculation — and the wrong answer, because the functional is blind to the very attraction that stabilizes the fold.
- B3LYP-D4 — the same calculation with a modern London-dispersion correction — flips the result: the folded conformer is now lower by 0.58 kcal/mol. Dispersion alone contributes −1.56 kcal/mol to the fold.
That is a 1.5 kcal/mol swing from a single missing physical effect — on a small, loosely-folded model. In a real diaryl drug with rings that stack closer, or a peptide macrocycle that collapses on itself, the dispersion term grows and the ranking error grows with it. A conformer search that stops at MMFF, or single-points at a dispersion-free functional, is quietly betting against London forces.
The receipts
| Model | E(folded − extended) | Says the winner is… |
|---|---|---|
| MMFF (force field) | +0.08 kcal/mol | a tie |
| B3LYP/def2-TZVP (no dispersion) | +0.98 kcal/mol | extended (wrong) |
| B3LYP-D4 (+ dispersion) | −0.58 kcal/mol | folded — the π-stacked form |
| … of which D4 dispersion | −1.56 kcal/mol | the force plain DFT missed |
Bibenzyl, 28 atoms. Single-point energies on the same MMFF folded (gauche, ~5.0 Å ring–ring) and extended (anti, ~6.5 Å) geometries; B3LYP/def2-TZVP with and without the D4 dispersion correction.
What this means for you: for any flexible molecule that can fold — diaryls, macrocycles, long linkers, anything with an intramolecular contact — rank conformers with a dispersion-aware method, not a bare force field or a dispersion-free functional. The bioactive conformation, the strain energy you subtract in a binding estimate, the shape your pharmacophore screen assumes: all of them sit on the conformer energy, and dispersion is often the term that decides the order.
The gold standard behind the correction
D4 is fast because it is calibrated: an atomic, charge-aware dispersion model fit to reproduce high-level reference data — ultimately coupled-cluster. When you want to check the correction rather than trust it, Hilbeon runs the explicit reference too: DLPNO-CCSD(T), the local-correlation “gold standard,” which captures dispersion from first principles rather than from a fitted formula. It is the same lesson as our H-bonds vs stacking study, where Hartree–Fock called two aromatic rings repulsive: binding, folding and stacking live in the correlation energy, and a method that ignores it will mislead you with a straight face.
Rank conformers with the right physics
Start a 30-day guided pilot — dispersion corrections, DFT and DLPNO-CCSD(T) included — and stop betting your conformer energies against London forces.
Honest edges
Three caveats, in the open. First, these are single-point energies on MMFF geometries: because a dispersion-bound fold is not a stationary point of a dispersion-free functional, comparing all methods on one consistent set of geometries is the fair test — and it isolates exactly the energy term we are probing. Second, bibenzyl is a deliberately small, loosely-folded model; the ~5 Å ring–ring contact makes this a conservative dispersion effect, and closer-stacking systems show more. Third, D4 is an empirical correction and — for bibenzyl specifically — the explicit DLPNO-CCSD(T) confirmation is our pending capstone rather than shown here; where the ranking is close or the money is on the line, that gold-standard reference is the arbiter, and the D4 result already agrees with the well-established dispersion physics of this system. None of that softens the headline: the same functional, plus or minus one physical term, changes which conformer it calls the winner.
References
- D4 dispersion — E. Caldeweyher et al., “A generally applicable atomic-charge dependent London dispersion correction”, J. Chem. Phys. 150, 154122 (2019).
- D3 dispersion — S. Grimme et al., J. Chem. Phys. 132, 154104 (2010).
- DLPNO-CCSD(T) local correlation — C. Riplinger & F. Neese, J. Chem. Phys. 138, 034106 (2013).