Methods deep-dive
Force Fields Can't See π-Stacking — Here's the Proof
Non-covalent interactions Binding Methods
Every so often someone asks the blunt question: does pharma actually need quantum chemistry? Docking is fast, force fields are cheap, and a good MM scoring function will rank a virtual library overnight. Here is the answer, and it is concrete. Drug binding is held together by weak, non-covalent forces. Take the two canonical ones — a hydrogen bond and a π-stack — and ask a simple thing: can the cheap methods that pharma leans on even get the sign right? We ran both complexes through Hilbeon's full HF → MP2 → CCSD → CCSD(T) hierarchy. One of them breaks the cheap methods completely.
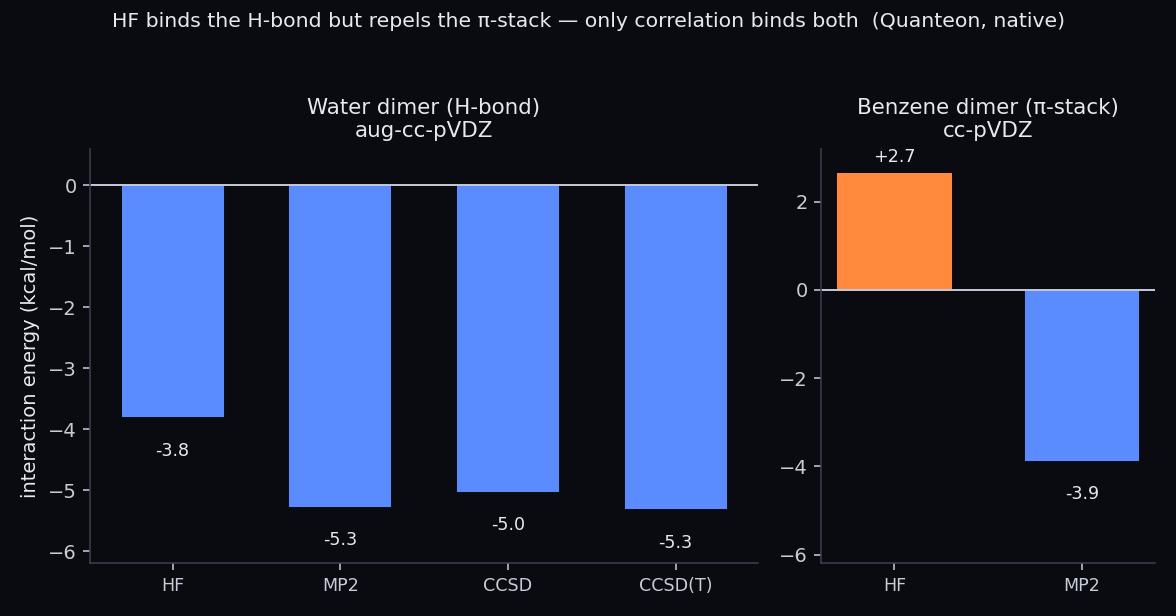
Interaction energies (Hilbeon). For the H-bond, HF captures most of the binding; for the π-stack, HF is repulsive (+2.7) — only correlation binds it. HF repels the stack; MP2 recovers the attraction.
Two complexes, two physics regimes
A non-covalent interaction energy is computed the supermolecular way: take the energy of the complex and subtract the energies of the two isolated monomers, ΔE = E(dimer) − E(A) − E(B). A negative number means the partners are bound. The two systems we picked are not arbitrary — they sit at opposite ends of what holds molecules together:
- The water dimer is the textbook hydrogen bond. Its binding is dominated by electrostatics: a polarized O–H donor pointing at a lone pair. That is permanent charge talking to permanent charge.
- The parallel-displaced benzene dimer is the textbook π-stack. There is no net charge, no permanent dipole, no classical electrostatic glue. What binds it is pure London dispersion — the instantaneous, correlated dance of electrons in one ring inducing a matching fluctuation in the other.
That second word, correlated, is the whole story. Dispersion is a correlation effect by definition. Hartree–Fock places each electron in the mean field of all the others and stops; it has no mechanism for two electron clouds to move in step. So we have a clean prediction before running anything: HF should do fine on the H-bond and should fail on the stack. Let's see how badly.
 Water dimer (hydrogen bond) — drag to rotate
Water dimer (hydrogen bond) — drag to rotateThe water dimer — one O–H donor pointing at the lone pair of the acceptor. A mostly electrostatic interaction.
The hydrogen bond: HF already gets most of it
Here is the full hierarchy for the water dimer at aug-cc-pVDZ — the diffuse basis you
want whenever lone pairs and weak interactions are involved:
| Method | Basis | ΔE (kcal/mol) | Note |
|---|---|---|---|
| HF | aug-cc-pVDZ | −3.82 | ~72% of CCSD(T) |
| MP2 | aug-cc-pVDZ | −5.29 | +correlation |
| CCSD | aug-cc-pVDZ | −5.04 | — |
| CCSD(T) | aug-cc-pVDZ | −5.33 | Hilbeon gold standard |
| DLPNO-CCSD(T0) | aug-cc-pVDZ | −5.21 | local method, 97.7% of our canonical CCSD(T) |
| CCSD(T)/CBS | reference | −5.0 | literature |
Read the first row. Hartree–Fock alone gives −3.82 kcal/mol — already bound, already about 72% of the CCSD(T) answer. That is the electrostatic backbone of the H-bond showing up exactly where theory says it should. Correlation then adds the remaining ~−1.5 kcal/mol: MP2 overshoots a touch to −5.29, CCSD pulls back to −5.04, and the gold-standard CCSD(T) settles at −5.33. For a hydrogen bond, the cheap method is wrong by a quarter, not by a category. A force field tuned to electrostatics and an explicit H-bond term can plausibly fake this.
The owned engine reaches the gold standard. Hilbeon's CCSD(T)/aug-cc-pVDZ water-dimer interaction energy (−5.33) lands right next to the published CCSD(T)/CBS reference (−5.0). That CCSD(T) value comes from Hilbeon's own coupled-cluster code on top of its own integral engine — no third-party libraries, fully deterministic and reproducible to the last digit. The full HF → MP2 → CCSD → CCSD(T) ladder is native. And the DLPNO row doubles as a validation: the local coupled-cluster engine, the one that scales to drug-sized molecules, reproduces 97.7% of the canonical interaction energy on the same dimer — the two implementations checking each other on the same physics.
 Benzene dimer (parallel-displaced π-stack) — drag to rotate
Benzene dimer (parallel-displaced π-stack) — drag to rotateThe parallel-displaced benzene dimer — two aromatic rings offset and stacked. No net charge, no permanent dipole: only dispersion holds it together.
The π-stack: where HF doesn't just miss — it flips the sign
Now the benzene dimer, the parallel-displaced geometry that dominates aromatic stacking in proteins and
DNA, at cc-pVDZ:
| Method | Basis | ΔE (kcal/mol) | Note |
|---|---|---|---|
| HF | cc-pVDZ | +2.68 | REPULSIVE — predicts no binding |
| MP2 | cc-pVDZ | −3.90 | binds it — correlation is the whole story (MP2 over-binds stacking) |
| DLPNO-CCSD | cc-pVDZ | −1.34 | coupled cluster without triples under-binds — (T) supplies the rest |
Look at the HF row and let it sink in. Hartree–Fock gives +2.68 kcal/mol — a positive number. HF does not merely underestimate the π-stack; it predicts that two benzene rings push each other apart. At the HF level the only forces in play are electrostatics and exchange repulsion, and with no permanent multipole to attract, all that is left is the Pauli wall. The result is a complex that, according to Hartree–Fock, should not exist. Any dispersion-less force field, and most docking scoring functions in their default form, make exactly the same mistake — they are built on the same electrostatics-plus-repulsion picture.
Switch on electron correlation and the physics reappears. MP2 swings all the way to −3.90 kcal/mol — the sign is now right, the stack binds. But MP2 famously over-binds π-systems; against the CCSD(T)/CBS reference of −2.65 it is too attractive by more than a kcal/mol. That over-binding is not a Hilbeon artifact — it is the well-documented failure mode of MP2 for aromatic stacking. Our own coupled-cluster row shows the other side of the same coin: DLPNO-CCSD gives −1.34 kcal/mol — without the perturbative triples, coupled cluster under-binds the stack by about as much as MP2 over-binds it. The two correlated rungs bracket the reference from opposite sides, which is exactly the textbook behaviour: the (T) triples supply the missing ~−1.3 kcal/mol and land on the gold-standard −2.65. One dispersion-bound complex, three rungs of the ladder, each failing — and self-diagnosing — in its own documented way.
Compute your own interaction energies
Bring a host–guest pair, a stacked dimer, an H-bonded motif — and run the whole HF → MP2 → CCSD → CCSD(T) ladder on it.
Reading the numbers honestly
These are real Hilbeon outputs, and we are going to be precise about what they do and don't prove.
What these numbers are — and aren't. These are supermolecular interaction energies computed without counterpoise correction: ghost-atom CP is a feature gap in the engine today. That means they carry basis-set superposition error (BSSE), which always pulls toward over-binding and is worst for MP2 with a small basis — so MP2's −3.90 on the benzene dimer is over-bound partly by genuine MP2 physics and partly by uncorrected BSSE. The bases here (cc-pVDZ, aug-cc-pVDZ) are also small, so on top of BSSE there is basis-set incompleteness. The upshot: treat the absolute magnitudes as approximate. What is robust is the sign and the qualitative conclusion — HF binds the H-bond and cannot bind the stack — which no amount of BSSE changes. For the stack we show our own HF (repulsive), MP2 (over-binding) and DLPNO-CCSD (under-binding) on the 24-atom dimer; the perturbative (T) triples on top — the rung that lands on the reference — carry a formal N⁷ cost we didn't run to completion here, but the full native HF→MP2→CCSD→CCSD(T) ladder on the water dimer above shows exactly how the triples close the gap. Geometries are fixed standard structures, not QM-reoptimized.
None of those caveats rescues Hartree–Fock. BSSE makes things more attractive, yet HF still lands at +2.68 on the stack. A larger basis and counterpoise would tighten the correlated numbers and shave MP2's over-binding, but they cannot turn a dispersion-less method into one that sees dispersion. The qualitative result — the part pharma actually has to act on — is the part that is bulletproof.
Why this is the answer to “does pharma need QM?”
π-stacking is not a corner case. It is everywhere a medicinal chemist looks: Phe / Tyr / Trp aromatic pockets, DNA and RNA base stacking, intercalators, the aromatic systems that pack against kinase hinge binders. Every one of those is held together by exactly the dispersion that Hartree–Fock, plain force fields, and default docking scores are constitutionally blind to. And the failure is not a 20% error you can calibrate around — it is a wrong sign, a prediction of repulsion where there is binding.
Here is the part that closes the loop. The force fields and docking functions that do capture π-stacking get it because someone added an explicit dispersion term — and that term was trained against correlated quantum chemistry — reference interaction energies at the CCSD(T)/complete-basis-set limit. The cheap methods are downstream of the expensive one. Correlated QM is not an optional luxury sitting next to docking; it is the benchmark that docking is calibrated to. When the calibration set is missing or the empirical term is off, the cheap method reverts to predicting that aromatic rings repel.
So: does pharma need quantum chemistry? For binding energetics, the honest answer is that pharma is already using it — second-hand, baked into the parameters — and reaches its limits the moment a new chemotype falls outside what those parameters were trained on. Hilbeon puts the full hierarchy directly in the chemist's hands: HF when electrostatics is enough, MP2 for a fast correlated estimate, and CCSD → CCSD(T) when you need the gold-standard number that everything else is measured against. That is why correlated QM is not optional for binding — and why it belongs in the toolkit, not just in the training set.
Run the gold standard on your own complex
From SMILES or a structure to CCSD(T) interaction energies — the same hierarchy, the same owned engine, your molecules.
References
- Aromatic π–π interactions — C. R. Martinez & B. L. Iverson, “Rethinking the term pi-stacking”, Chem. Sci. 2012, 3, 2191
- S66 extended benchmark set — Řezáč, Riley, Hobza, “S66: A Well-balanced Database of Benchmark Interaction Energies”, J. Chem. Theory Comput. 2011, 7, 2427
- Benzene-dimer CCSD(T)/CBS — Sinnokrot & Sherrill, “Highly Accurate Coupled Cluster Potential Energy Curves for the Benzene Dimer”, J. Phys. Chem. A 2004, 108, 10200
- MP2 over-binding of π-stacking — Sinnokrot & Sherrill, “Substituent Effects in π–π Interactions”, J. Am. Chem. Soc. 2004, 126, 7690
- Dispersion in drug binding — Bissantz, Kuhn, Stahl, “A Medicinal Chemist's Guide to Molecular Interactions”, J. Med. Chem. 2010, 53, 5061