Application note
Seeing the Forces: an NCI Portrait of Salicylic Acid
NCI Drug discovery Permeability
The structure drawing you sketch on a whiteboard is a lie of omission. It shows the covalent bonds — the scaffolding — but hides the weak forces that actually decide whether a molecule binds, dissolves and crosses a membrane. Non-covalent interactions (NCI) analysis fixes that: it lets you see hydrogen bonds, van der Waals contacts and steric clashes directly, painted onto the real-space electron density. Our subject is salicylic acid — aspirin's active metabolite — which hides a textbook intramolecular hydrogen bond in plain sight. Every number below was computed in Hilbeon: the 3D structure comes from RDKit (ETKDG + MMFF), which Hilbeon then geometry-optimizes; the numbers come from a B3LYP/6-31G(d) calculation and an NCI analysis on that optimized minimum.
 Salicylic acid — drag to rotate
Salicylic acid — drag to rotate
Salicylic acid in 3D — drag to rotate. The phenol –OH and the carboxylic acid sit next door to each other on the ring; that proximity is the whole story.
Why salicylic acid? Aspirin, finished
If you read our computational portrait of aspirin, you already met this molecule as a plot twist. Aspirin is its own prodrug: the acetyl ester hydrolyses in the body to release salicylic acid, the species that actually does much of the work. So this is not a detour — it is the sequel. Where aspirin's story ended with a vulnerable ester, salicylic acid's begins with what the ester unmasks: a phenol –OH sitting one bond away from a carboxylic acid, perfectly placed to fold back and grab it.
That fold-back is an intramolecular hydrogen bond: the phenol O–H donates to the carboxyl C=O acceptor at an O···H distance of just 1.80 Å, closing a six-membered ring of atoms and locking the molecule planar (0.002 Å from its best-fit plane). You can guess it exists from the structure. NCI lets you stop guessing and look at it.
What NCI actually shows you
NCI is built on a simple, beautiful observation. The reduced density gradient (RDG) — a dimensionless measure of how fast the electron density is changing — drops toward zero not only inside covalent bonds, but also in the low-density gaps between non-bonded fragments where a weak interaction lives. Plot RDG against the density and those weak-interaction regions show up as distinct spikes. To tell an attractive contact from a repulsive one, NCI signs the density by the sign of the second Hessian eigenvalue, sign(λ₂)ρ: negative means attractive (a hydrogen bond), positive means steric repulsion, near-zero means a soft van der Waals contact. Map that back onto 3D space and you get a coloured surface — blue for attractive, green for weak vdW, red for steric — that floats exactly where the forces act.
Crucially, none of this needs you to define a bond, pick atoms, or assume a hydrogen bond is there. It falls out of the density itself. Hilbeon computes the density on a fine grid (here, 0.12 bohr spacing), evaluates the RDG and the signed density, and hands you both the 3D surface and the 2D diagnostic plot.
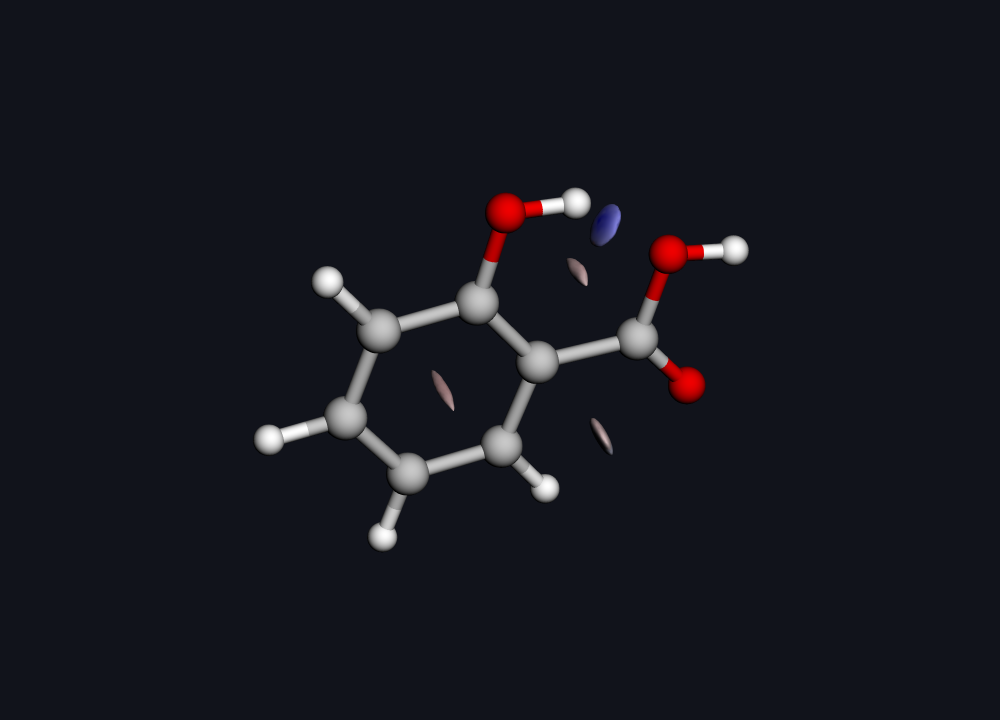
Real Hilbeon NCI surface. Blue disk between the phenol O-H and the carboxyl C=O = the attractive intramolecular hydrogen bond; the reddish patch in the ring centre = steric repulsion.
There it is. The flat blue disk wedged between the phenol hydrogen and the carboxyl oxygen is the hydrogen bond — an attractive interaction, drawn straight from the electrons, no prior assumption required. The reddish patch hovering over the ring centre is the unavoidable steric repulsion of a closed aromatic system. One picture, both characters of force, in their true positions.
The fingerprint: reading the 2D plot
The 3D surface is the poster; the 2D plot is the lab notebook. It is the canonical NCI fingerprint — RDG on the vertical axis, sign(λ₂)ρ on the horizontal — and you read it left to right. A trough on the left (negative side) is an attractive interaction; the further left, the stronger. A spike on the right (positive side) is steric strain.
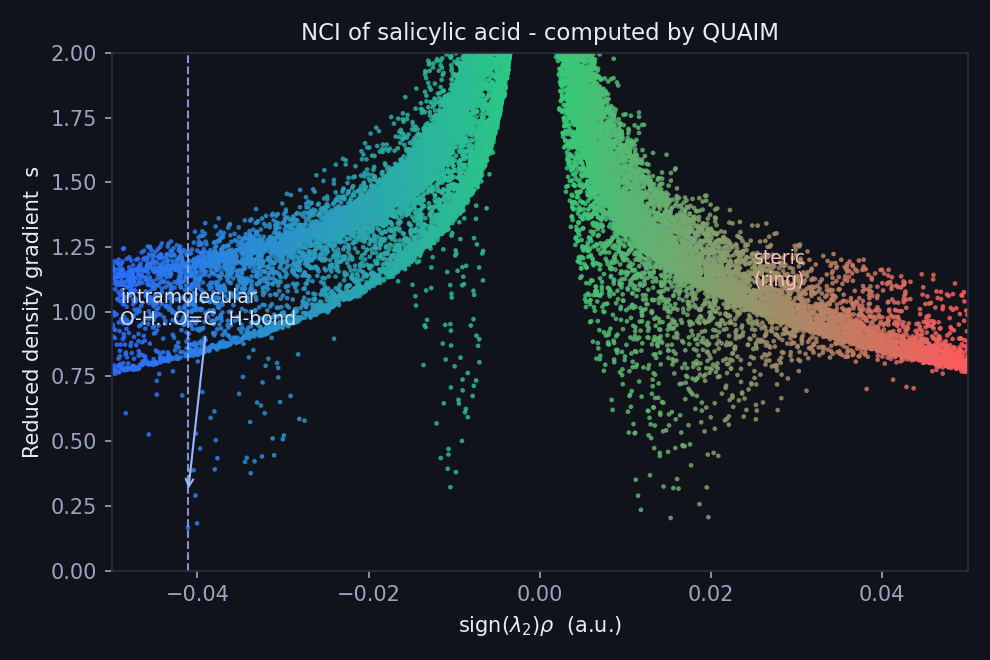
The classic NCI fingerprint (RDG vs sign(λ2)ρ). The spike at sign(λ2)ρ ≈ −0.041 a.u. is the hydrogen bond; the right-hand red wing is steric.
For salicylic acid the attractive trough sits at sign(λ₂)ρ ≈ −0.041 a.u., with the reduced density gradient on the attractive branch dipping to a minimum of 0.166 there. That position is the diagnostic: a value around −0.04 a.u. is the signature of a moderately strong hydrogen bond — well to the left of a weak van der Waals contact (which would huddle near zero), but not as deep as the very strongest ionic H-bonds. In plain terms: this is a real, structure-defining interaction, not a casual brush of atoms. The red wing on the right is the ring's steric crowding, exactly as the 3D surface promised.
The chameleon trick: a masked dipole
Here is where the chemistry pays off. A phenol and a carboxylic acid are both polar groups. Left to point outward, they would give the molecule a hefty dipole and make it eager to stay in water — bad news for crossing a fatty membrane. But the intramolecular hydrogen bond folds those two polar groups toward each other, so their electrostatics partly cancel. The molecule effectively tucks its polarity inside.
Hilbeon puts a number on it. The QM dipole of salicylic acid in its folded, H-bonded form is just 0.72 D — against aspirin's 2.00 D from the same engine. And this fold is no fleeting shape: it is the global minimum, 7.5 kcal/mol below the open rotamer (whose dipole is a hefty 3.35 D). That near-threefold drop is not because salicylic acid is less polar in its parts; it is because the H-bond masks the polarity. This is the "molecular chameleon" behaviour that medicinal chemists deliberately exploit: a molecule that looks polar on paper but presents a low-polarity, membrane-friendly face when it folds. It is one of the central tactics for pushing larger, polar, "beyond-Rule-of-5" molecules across membranes — and an intramolecular hydrogen bond is the most common way to do it.
The supporting cast confirms the assignment. The NCI reading and the near-planar geometry place the phenol O–H proton as the hydrogen-bond donor and the carboxyl C=O oxygen as the acceptor — the same two atoms the NCI blue disk sits between. The frontier electronics are unremarkable and healthy: a HOMO at −8.78 eV (RHF/6-31G(d)), consistent with a stable closed-shell aromatic acid. The point of this molecule was never its orbitals; it was its shape, and the force that holds it.
See your molecule's hidden interactions
From SMILES to an NCI surface in one command. If your compound has an intramolecular H-bond hiding its polarity, Hilbeon will show you exactly where.
Honesty check. NCI shows the presence and character of an interaction — attractive hydrogen bond versus steric clash — not a numeric bond energy. The position of the trough (−0.041 a.u. here) ranks strength qualitatively; it is not a ΔG. The geometry is now QM-optimized (B3LYP/6-31G(d)), and the folded, H-bonded form is the global minimum — 7.5 kcal/mol below the open rotamer — so it genuinely dominates rather than being one shape among many. The masked dipole is the property of that dominant fold; a molecule still samples other shapes in solution, so quantify with conformer sampling when a precise number is needed. Treat NCI as a sharp diagnostic of what interaction is present and where, then quantify with conformer sampling and free-energy methods when a number is needed.
The receipts: what Hilbeon computed
Here is the honest scorecard. Every value in the Hilbeon column is a real number from the run that produced the NCI surface above — a B3LYP/6-31G(d) calculation and an NCI analysis on the QM-optimized, H-bonded geometry.
| Property | Hilbeon computed | Reference / note | Source |
|---|---|---|---|
| Total energy — RHF/6-31G(d) | −493.1728 Ha | on the QM-optimized geometry | Hilbeon |
| HOMO — RHF/6-31G(d) | −8.78 eV | stable closed-shell aromatic acid | Hilbeon |
| Dipole moment (QM) | 0.72 D | vs aspirin 2.00 D — the H-bond masks polarity | Hilbeon |
| NCI hydrogen-bond trough | sign(λ₂)ρ ≈ −0.041 a.u. | moderately strong attractive H-bond | Hilbeon NCI |
| NCI minimum RDG (attractive) | 0.166 | character, not a bond energy | Hilbeon NCI |
| H-bond donor / acceptor | phenol O–H / carboxyl C=O | from the NCI reading and near-planar geometry | Hilbeon NCI |
Scope note. The energy and HOMO above are bench-grade electronic-structure numbers. The NCI surface, the 2D fingerprint and the donor/acceptor reading are insight and visualization — they tell you what interaction is present and where it sits, not a single bench-matched value. Hilbeon's integral engine is its own (McMurchie–Davidson + Rys, no third-party libraries), GPU-accelerated, and fully deterministic — bit-identical across machines.
The whole portrait, in four prompts
Every figure above came from talking to the Hilbeon assistant in plain language — no input decks, no NCI grid syntax to memorize:
build salicylic acid from SMILES O=C(O)c1ccccc1O
optimize the geometry at B3LYP/6-31G(d)
compute the dipole moment
run an NCI analysis and render the surface and the 2D plotThat is the entire workflow — from SMILES to a hydrogen bond you can see. Prefer scripts? Each step is also a call in Hilbeon's MCP / HTTP API, so the whole portrait is reproducible end-to-end in a notebook or a CI job.
The takeaway
Salicylic acid's electrons told us something the structure drawing could not: a single intramolecular hydrogen bond, sitting at sign(λ₂)ρ ≈ −0.041 a.u. on the NCI plot, folds two polar groups into each other and drops the dipole from 2.00 D to 0.72 D. That is the molecular-chameleon trick — visible, located, and characterised straight from the density. The same NCI analysis is one prompt away for whatever is on your bench.
See your molecule's hidden interactions
Start a 30-day guided pilot — every method, every core, your GPU — and render an NCI portrait of the compound you actually care about.
References
- NCI method — Johnson et al., J. Am. Chem. Soc. 2010; overview at Non-covalent interactions index
- Intramolecular H-bonds & permeability — Caron et al., Med. Res. Rev. 2019 (beyond-Rule-of-5 review)
- Salicylic acid — PubChem: Salicylic acid (CID 338)
- Aspirin as a prodrug — A Computational Portrait of Aspirin