Application note
Switching a Drug On with Light: Azobenzene by TD-DFT
Photopharmacology Excited states Drug design
Most drugs are “always on” everywhere they reach — which is precisely why side effects happen. Photopharmacology builds a light-operated switch into the molecule: shine one colour of light and the drug folds into its active shape; shine another, or simply wait, and it relaxes back to an inactive one. Activate the tumour, not the healthy tissue around it. The molecular workhorse for that trick is azobenzene — a Ph–N=N–Ph scaffold whose –N=N– hinge flips between a flat trans and a bent cis under illumination.
The entire design hinges on one question: what colour of light flips it? That is not a ground-state question you can read off a static structure — it is an excited-state question, exactly the kind that time-dependent DFT answers. In this post we point Hilbeon's TD-DFT at azobenzene and watch the textbook photoswitch fall straight out of the wavefunction: two isomers, two spectra, the energy the photon banks in the bent shape, a triplet channel into singlet-oxygen chemistry, and a concrete lever for moving the switch into the tissue-penetrating window. From a SMILES string to a predicted absorption spectrum and a singlet–triplet gap, in one run, on a workstation — no HPC cluster.
The two shapes: a flat trans and a bent cis
Azobenzene comes in two geometries that interconvert across the –N=N– double bond. The stable trans (E) isomer is essentially planar: Hilbeon's geometry optimization puts its C–N=N–C dihedral at 179.9°, the two phenyl rings stretched out flat across the azo bridge. Absorb the right photon and the molecule rotates/inverts about that bridge into the metastable cis (Z) isomer, which is sharply bent — the CNNC dihedral collapses to 9.8°, folding the two rings onto the same side.
Optimized trans-azobenzene (B3LYP/6-31G(d), geodesic RIC-RFO). The molecule is planar — CNNC = 179.9° — with the two phenyl rings extended across the –N=N– bridge. Drag to rotate.
Optimized cis-azobenzene (B3LYP/6-31G(d), geodesic RIC-RFO). CNNC = 9.8°: the rings fold onto the same side of the bent azo bridge. Drag to rotate.
The cis isomer sits uphill of the trans — that difference is the energy a photon banks when it flips the switch, and it is what makes the stored-energy number worth getting right. Our first attempt at B3LYP/6-31G(d) gave a cis–trans gap of +15.21 kcal/mol, well above the experimental band of roughly 10–12 kcal/mol. The instinct is to blame the basis set — so we went to def2-TZVP and the gap barely moved, to +15.12 kcal/mol. The basis was already converged; the error was physics, not size. The compact cis isomer, with its two rings on the same side, is stabilized by intramolecular dispersion that a global hybrid omits. Adding a D4 dispersion correction pulls the gap down to +12.94 kcal/mol at 6-31G(d) and +12.84 kcal/mol at def2-TZVP — at the upper edge of the experimental band (and the ~0.5–1 kcal/mol of zero-point energy we have not added would bring it down further). The lesson is a clean teaching point: here it was dispersion, not basis.
How these geometries were found. Both isomers were optimized with Hilbeon's geodesic RIC-RFO optimizer (internal coordinates + rational-function-step + model Hessian + GEDIIS + geodesic stepping), B3LYP/6-31G(d), converged on max force below 4.5×10−4 Eh/Å. Trans converged in 9 steps, cis in 19. This matters: the legacy Cartesian BFGS optimizer did not converge azobenzene at all — it oscillated for 41+ steps on the floppy phenyl torsions. These are genuine QM minima, not force-field guesses.
The colour that flips it
Now the excited states. Azobenzene's UV-Vis is governed by two transitions: a weak, low-energy n→π* band (the nitrogen lone pair into the azo π*) and an intense, higher-energy π→π* band. The whole photoswitch lives in how those two bands move and change intensity between the isomers. Hilbeon's TD-DFT (full Casida, Davidson solver) reproduces the textbook picture, and the simulated spectra below make the contrast visible at a glance.
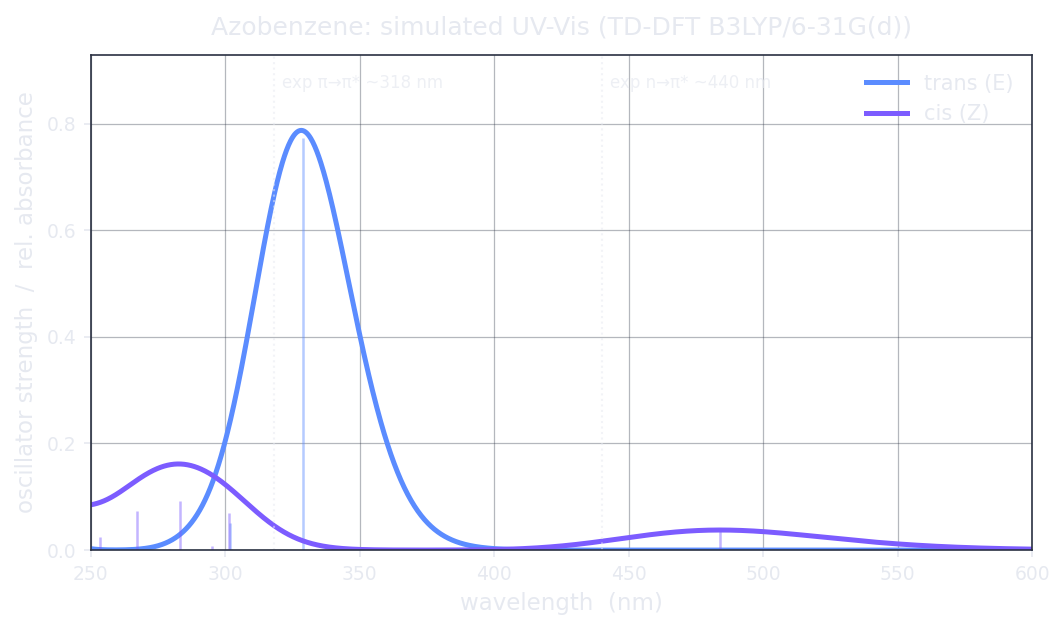
Simulated UV-Vis of trans vs cis azobenzene (TD-DFT B3LYP/6-31G(d), Gaussian-broadened vertical excitations, σ = 0.20 eV). The bright π→π* band dominates the planar trans spectrum; bending to cis switches the n→π* band on and blue-shifts the π→π*.
| Isomer / band | Energy (eV) | λ (nm) | Oscillator strength f | Reference |
|---|---|---|---|---|
| trans — n→π* | 2.54 | 488.3 | ~1×10−6 (dark) | exp ~440–460 nma; Hilbeon is ~0.28 eV too low (the known B3LYP n→π* error) |
| trans — π→π* (bright) | 3.77 | 328.8 | 0.7736 | exp ~318 nma; within ~11 nm |
| cis — n→π* (switched on) | 2.56 | 484.0 | 0.0376 | band is allowed (exp ~440 nma); Hilbeon is ~0.26 eV too low on the wavelength — the qualitative switch-on is the robust result |
| cis — π→π* (blue-shifted) | 4.38 | 283.1 | 0.093 | exp ~280 nma; within ~3 nm |
a Experimental band positions for cis/trans azobenzene from H. M. D. Bandara & S. C. Burdette, Chem. Soc. Rev. 2012, 41, 1809. The valence π→π* positions agree to within a few nm; the n→π* bands sit ~0.26–0.28 eV too low — the textbook B3LYP underestimation of the azobenzene n→π* energy. The computed columns (energy, λ, f) are Hilbeon TD-DFT output; the experimental values are literature references shown for context.
Read the table and the switch explains itself. In planar trans the n→π* is essentially dark — f ≈ 10−6, symmetry-forbidden in the flat geometry, so that near-zero vertical f is partly a Franck–Condon artifact (the band is seen weakly in experiment through vibronic borrowing) — while the π→π* is intense at 328.8 nm with f = 0.77 (against an experimental ~318 nm). Bending to cis breaks that symmetry: the n→π* band switches on to f = 0.038 — a factor of tens of thousands brighter — while the π→π* blue-shifts all the way to 283.1 nm. That intensity-and-wavelength difference between the two isomers is the whole game: it means you can shine a colour that one isomer absorbs and the other barely does, addressing one form without fully exciting the other. That selective addressing is reversible photoswitching.
Which orbitals do the work
The two bands are two different electronic motions, and naming them is the key to the photochemistry. The n→π* excitation lifts an electron out of a nitrogen lone pair — the “n” orbital localised on the azo nitrogens, lying in the molecular plane — into the antibonding π* of the N=N bridge. Because that lone pair is essentially orthogonal to the π system in the planar trans isomer, the transition is near-forbidden and weak; bending to cis tilts the geometry, mixes the orbitals, and lights it up. The π→π* excitation, by contrast, promotes an electron within the conjugated π framework spanning both rings and the azo bridge — a fully allowed, intense transition, the strong band in the trans spectrum.
That orbital character is also why the photon does mechanical work: populating the azo π* weakens the N=N π bond and lets the molecule rotate or invert about it, carrying the system from one isomer minimum toward the other. A cleaner way to visualise exactly which orbitals participate would be natural transition orbitals (NTOs), which collapse each excitation into a single particle–hole pair — a refinement we flag as the natural next step for a per-state picture rather than reasoning from the canonical frontier orbitals alone.
A triplet channel into singlet-oxygen chemistry
Excited states are not only singlets. Hilbeon's triplet TD-DFT places azobenzene's lowest triplet T1 at 1.76 eV in the trans isomer and 1.55 eV in the cis, with singlet–triplet (S1–T1) gaps of 0.778 eV and 1.011 eV respectively. The triplet matters for two reasons. First, it opens an alternative, sensitized switching channel: a triplet photosensitizer can hand energy to azobenzene and drive isomerization with lower-energy light than a direct singlet excitation would need. Second, a populated triplet near ~1.6–1.8 eV is the gateway to photodynamic chemistry — energy transfer from a long-lived triplet to ground-state molecular oxygen generates cytotoxic singlet oxygen, the active agent in photodynamic therapy. The same one excited-state run that gives you the colour gives you these triplet handles for free.
Red-shifting the switch toward the clinic
There is a catch for real photopharmacology: the bands above are addressed with UV light, which is phototoxic and barely penetrates tissue. To flip a switch inside a patient you want to drive it with red or near-infrared light in the tissue-transparency window. The classic lever is a push–pull azobenzene — install an electron donor on one ring and an electron acceptor on the other. We built the 4-NH2 / 4′-NO2 derivative and ran the same TD-DFT.
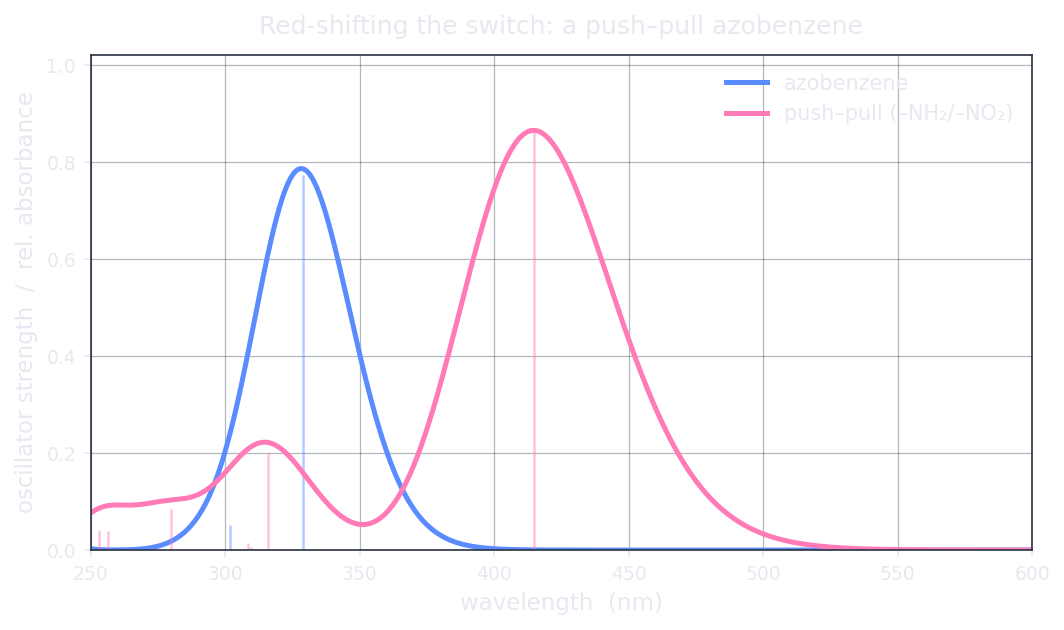
Plain azobenzene (bright band 328.8 nm) vs the push–pull 4-NH2/4′-NO2 derivative (bright band 414.7 nm). The donor–acceptor pair drags the absorption out of the UV and into the visible — the direction a photopharmacologist wants.
The push–pull azobenzene: an electron-donating amino group on one ring, an electron-withdrawing nitro on the other. The asymmetry sets up a donor-to-acceptor charge-transfer transition. Drag to rotate.
It works as intended. The bright band moves from 328.8 nm in plain azobenzene to 414.7 nm (2.99 eV, f = 0.866) in the push–pull system — a clean march out of the UV and into the visible. The donor–acceptor substitution does exactly what photopharmacology asks of it: it shifts the operating colour toward the tissue window.
An honest caveat on the magnitude. That red-shifted band is a charge-transfer (CT) excitation — an electron moving from the amino donor to the nitro acceptor across the molecule. Global hybrid functionals like B3LYP systematically underestimate CT excitation energies, which means the true band may sit at a somewhat different wavelength than the 414.7 nm we report. The direction of the result — donor/acceptor substitution red-shifts the bright band into the visible — is robust and well-established; the quantitative magnitude needs a range-separated hybrid (CAM-B3LYP or ωB97X), which is on Hilbeon's roadmap. We show you the lever and we name its limit.
Read the colour of your own photoswitch
From a SMILES string to a predicted absorption spectrum and a singlet–triplet gap in one excited-state run, on a workstation — no HPC cluster. Point Hilbeon's TD-DFT at your chromophore.
The receipts: what Hilbeon computed
Here is the honest scorecard. Every excitation energy, wavelength and oscillator strength below is a real number from a Hilbeon TD-DFT run — full Casida equations, Davidson solver — at B3LYP/6-31G(d) on geometries optimized at the same level with the geodesic RIC-RFO optimizer. The stored energy is the refined B3LYP-D4/def2-TZVP electronic value.
| Quantity | Hilbeon computed | Reference / note |
|---|---|---|
| trans bright π→π* | 3.77 eV / 328.8 nm / f 0.7736 | exp ~318 nm (Bandara & Burdette 2012); within ~11 nm |
| trans n→π* | 2.54 eV / 488.3 nm / f ~1e−6 | dark in the planar isomer; exp ~440–460 nm — Hilbeon ~0.28 eV too low (B3LYP n→π* error) |
| cis n→π* | 2.56 eV / 484.0 nm / f 0.0376 | switched on by bending; exp ~440 nm — Hilbeon ~0.26 eV too low |
| cis π→π* | 4.38 eV / 283.1 nm / f 0.093 | blue-shifted vs trans; exp ~280 nm; within ~3 nm |
| push–pull bright band (CT) | 2.99 eV / 414.7 nm / f 0.866 | UV→visible; B3LYP underestimates CT magnitude |
| T1 (trans / cis) | 1.76 / 1.55 eV | triplet TD-DFT |
| S1–T1 gap (trans / cis) | 0.778 / 1.011 eV | sensitization / singlet-oxygen channel |
| Stored energy (cis − trans) | +12.84 kcal/mol | B3LYP-D4/def2-TZVP; exp ~10–12 |
Scope of these numbers. They are vertical (Franck–Condon) excitation energies, not band maxima with vibronic structure — the simulated spectra are Gaussian-broadened sticks (σ = 0.20 eV), so the positions are the result and the band shape is illustrative. They are gas-phase; experimental UV-Vis is in solution and the n→π*/CT bands are solvatochromic (state-specific PCM on excited states is a roadmap gap). B3LYP is excellent for the valence n→π*/π→π* of cis/trans azobenzene but underestimates the push–pull charge-transfer magnitude — a range-separated hybrid is needed there. The stored energy is electronic (no ZPE/thermal added, ~0.5–1 kcal/mol, within the experimental scatter). Geometries are genuine QM minima from the geodesic RIC-RFO optimizer, not force-field structures.
Computed on our own engine. Hilbeon runs on its own integral and Casida engine — no third-party libraries in the hot path — and every result is fully deterministic, reproducible to the last digit across machines. The numbers here are Hilbeon's own output straight from the molecule's electron density, not a fitted model or a borrowed value: the trans bright π→π* at 3.771 eV (f 0.774) and T1 at 1.761 eV. The spectra you are reading are the real ones.
How to do it yourself
Everything above came from talking to the Hilbeon assistant in plain language — no input decks, no excited-state syntax to memorize:
build trans-azobenzene from SMILES c1ccc(cc1)/N=N/c1ccccc1
optimize the geometry at B3LYP/6-31G(d)
run TD-DFT B3LYP/6-31G(d): 12 singlets and 3 triplets
report each state's energy, wavelength and oscillator strength
simulate the broadened UV-Vis spectrum
repeat for the cis isomer (.../N=N\c1ccccc1) and compare
build the push-pull analogue Nc1ccc(cc1)/N=N/c1ccc(cc1)[N+](=O)[O-] and see the red-shiftPrefer scripts? Every one of those steps is also a call in Hilbeon's MCP / HTTP API, so sweeping a library of candidate switches is a loop in a notebook. The full excited-state workup — optimization, singlets, triplets, broadened spectrum — runs end to end on a single workstation, no cluster queue.
The takeaway
Azobenzene's photoswitch is an excited-state story, and Hilbeon reads it straight off the wavefunction: a flat trans and a bent cis, separated by +12.8 kcal/mol of dispersion-stabilised stored energy; a dark trans n→π* that switches on in the cis; a bright π→π* that blue-shifts on bending — the intensity-and-wavelength contrast that makes reversible switching possible; a triplet channel into photodynamic chemistry; and a push–pull lever that drags the operating colour toward the tissue window. From SMILES to spectrum to singlet–triplet gap, in one run. Now point the same engine at the chromophore on your bench — and find out what colour flips it before you commit a synthesis.
Read the colour of your own photoswitch
Start a 30-day guided pilot — every method, every core, your GPU — and run the full excited-state workup on the compound you actually care about.
References
- Photopharmacology — W. A. Velema, W. Szymanski & B. L. Feringa, J. Am. Chem. Soc. 2014, 136, 2178
- Azobenzene photoswitches, a review — H. M. D. Bandara & S. C. Burdette, Chem. Soc. Rev. 2012, 41, 1809
- Photoswitches in biology — M. M. Lerch, M. J. Hansen, G. M. van Dam, W. Szymanski & B. L. Feringa, Angew. Chem. Int. Ed. 2016, 55, 10978
- TD-DFT of azobenzene excited states — L. M. Frutos et al.; and the Casida formalism, TD-DFT (overview)
- Charge-transfer failure of global hybrids — A. Dreuw & M. Head-Gordon, Chem. Rev. 2005, 105, 4009
- Optimizing on excited states — analytic excited-state gradients in Hilbeon