In the literature
Inside Orforglipron: A 113-Atom Drug by Quantum Chemistry
Drug portrait Frontier orbitals Scale
In April 2026 the FDA approved orforglipron (Foundayo) — the first oral, non-peptide GLP-1 receptor agonist, the long-sought “GLP-1 in a pill.” It is a small molecule that mimics what a peptide hormone does at its receptor, and it is big for a small molecule: Cₜ₈Hₜ₈F₂N₁₀O₅, molar mass 883, 65 heavy atoms. This post does something simple and, for a molecule this size, not entirely trivial: it builds orforglipron from its SMILES string and converges a real ab-initio wavefunction of it — then reads what the electrons are doing.
The molecule
Orforglipron's architecture is a far cry from a textbook drug. A central biaryl-azole core, two fluorinated rings, a fused triazole, an oxadiazolone, a spirocyclic tetrahydropyran — the kind of three-dimensional, sp³-rich scaffolding modern medicinal chemistry uses to fold a small molecule into the shape of a peptide. Drag the structure below: 113 atoms, every one placed by Hilbeon from the SMILES.
Orforglipron, Cₜ₈Hₜ₈F₂N₁₀O₅ (PubChem CID 137319706), built from SMILES with RDKit (ETKDG + MMFF). 113 atoms, 464 electrons. Drag to rotate.
From SMILES to a converged wavefunction
Building a 3D structure is easy; converging quantum mechanics on it is where big, floppy drug molecules fight back. Hilbeon ran a Hartree–Fock calculation in the 6-31G(d) basis — 1006 basis functions spanning 464 electrons — in integral-direct mode, and it converged cleanly to a total energy of −2963.1513937597 Ha. We checked the same calculation on two different machines (a workstation and a laptop, different builds): bit-for-bit identical to the last digit.
This one was a genuinely hard SCF. A molecule this large and conformationally soft has densely packed, near-degenerate frontier orbitals, and a naive starting guess sends the iteration sliding into a bad basin where the energy diverges by thousands of Hartree per cycle. Getting it to converge — deterministically, and identically across machines — takes a well-conditioned initial guess and a robust accelerator. That it lands on the same number on two builds is the quiet, important part: reproducible quantum chemistry on a real drug, not just on water.
What the electrons are doing
With a converged wavefunction in hand, the frontier orbitals are the first thing a chemist reads — they are where reactivity and metabolism live.
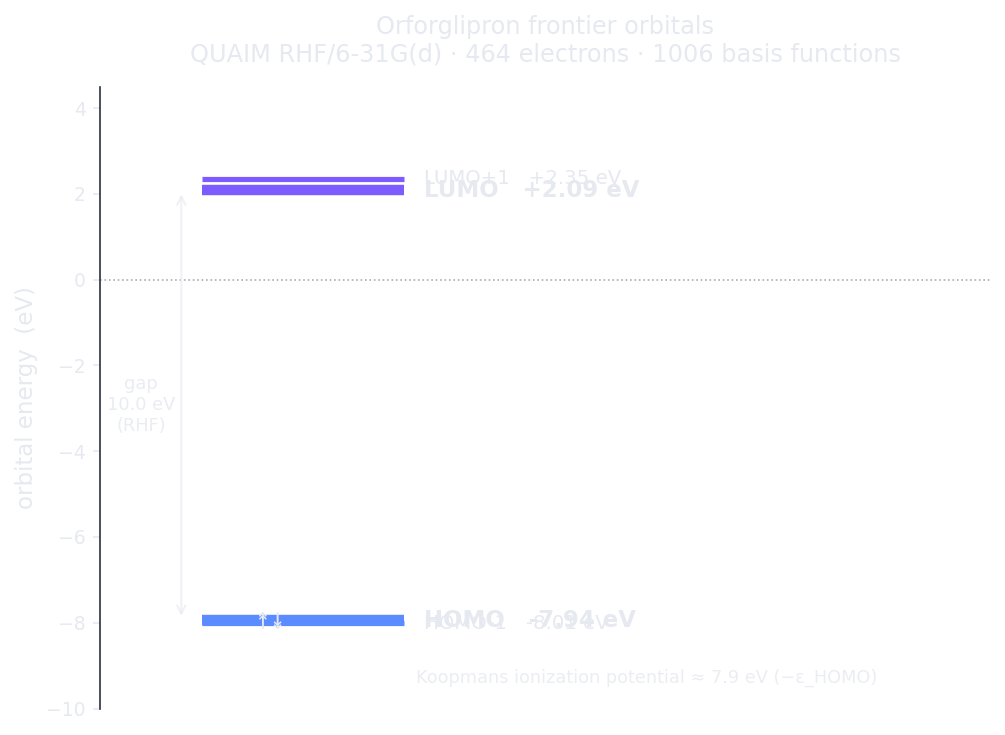
Orforglipron's frontier orbitals, Hilbeon RHF/6-31G(d). The HOMO sits at −7.94 eV; by Koopmans' theorem that is an ionization potential of ≈ 7.9 eV.
The HOMO at −7.94 eV is the most useful single number here. Read as a Koopmans ionization potential (−εHOMO ≈ 7.9 eV), it measures how tightly the molecule holds its most loosely-bound electrons — a first-principles handle on the molecule's electron-donating capacity and its susceptibility to oxidative metabolism, exactly the kind of property that decides a drug's metabolic stability. The LUMO at +2.09 eV marks the lowest unoccupied level. These are physical orbital energies — the kind a frontier-orbital argument actually rests on.
| Quantity | Hilbeon RHF/6-31G(d) | Note |
|---|---|---|
| Total energy | −2963.1513937597 Ha | converged; bit-identical on two machines |
| HOMO | −7.94 eV | Koopmans IP ≈ 7.9 eV |
| LUMO | +2.09 eV | — |
| HOMO–LUMO gap | 10.0 eV | RHF (overestimates the optical gap) |
| Size | 113 atoms · 464 e⁻ · 1006 basis fns | integral-direct |
SMILES: Cc1cc(-n2nc3c(c2-n2ccn(-c4ccc5c(cnn5C)c4F)c2=O)[C@H](C)N(C(=O)c2cc4cc([C@H]5CCOC(C)(C)C5)ccc4n2[C@@]2(c4noc(=O)[nH]4)C[C@@H]2C)CC3)cc(C)c1F. Geometry from RDKit ETKDG + MMFF; energies fully deterministic and reproducible to the last digit across machines.
What this is — and is not. This is a single-point calculation on an RDKit/MMFF geometry, not a QM-optimized structure — orforglipron is large enough that a full geometry optimization is a workstation/cluster job, not a laptop one. The energies are Hartree–Fock: the HOMO and the ionization potential are physically meaningful, but Hartree–Fock systematically overestimates the HOMO–LUMO gap, so the 10 eV gap is not the molecule's optical gap (a DFT or excited-state calculation would give a realistic absorption energy). Read this as a faithful first-principles first look at a landmark molecule — the frontier-orbital topology and the ionization potential — not as a finished property dossier.
Put your own molecule under the same lens
From a SMILES string to a converged ab-initio wavefunction of a 113-atom drug, on a workstation — no HPC cluster. Hilbeon scales to the molecules you actually work on.
Why “it converged” is the headline
It is easy to take SCF convergence for granted on small molecules and forget that it is not guaranteed. On a 65-heavy-atom drug with a near-degenerate frontier, a quantum chemistry package either gives you a reproducible number or it slides off into nonsense — and which one you get can depend on the machine and the build. The point of this portrait is that Hilbeon gives the same converged wavefunction of orforglipron on a laptop and on a workstation. That reproducibility — on a real, awkward, drug-sized molecule — is what makes the frontier-orbital numbers above worth trusting.
How to do it yourself
orforglipron now ships in Hilbeon's molecule set — pick it and run, in plain language:
load orforglipron
run Hartree-Fock in 6-31G(d), integral-direct
report the HOMO, the LUMO and the ionization potential
(then try DFT/B3LYP for a realistic optical gap)The takeaway
Orforglipron is a milestone in medicinal chemistry — a peptide hormone's job, done by a pill. It is also a big, stubborn molecule for first-principles quantum chemistry. Hilbeon builds it from a SMILES string, converges a real Hartree–Fock wavefunction of all 1006 basis functions, and returns the same answer on two machines, with physical frontier orbitals to read. From a string to a wavefunction, on hardware you already own — now point it at the molecule on your bench.
Bring the quantum chemistry in-house
Start a 30-day guided pilot — every method, every core, your GPU — and run the molecule you actually care about, however big.
References
- Orforglipron — structure and identifiers, PubChem CID 137319706
- Orforglipron (oral non-peptide GLP-1 receptor agonist) — overview, Wikipedia
- Koopmans' theorem (HOMO energy as ionization potential) — T. Koopmans, Physica 1934, 1, 104
- The engine that computes it — the Hilbeon blog