In the literature
The Quantum Chemistry Behind the 2025 Nobel: How MOFs Grab CO₂
Carbon capture MOFs Reaction energetics
The 2025 Nobel Prize in Chemistry went to Susumu Kitagawa, Richard Robson and Omar Yaghi for metal–organic frameworks — crystalline scaffolds, riddled with molecular-sized cavities, that can pull CO₂ straight out of a gas stream or wring water from desert air. A MOF is a vast, periodic framework, far beyond what a single-molecule engine should ever try to simulate. But the chemistry that actually grabs the CO₂ happens at one molecular site — the appended amine — and that reaction is squarely in Hilbeon's wheelhouse.
So this post does not pretend to compute a framework. It zooms in on the binding-site reaction the prize is built on, runs it in Hilbeon, and watches a subtle, well-documented experimental result fall out of the energetics: in dry conditions the amine and CO₂ form a neutral carbamic acid, but add water and the product switches to an ionic ammonium carbamate. From a reaction you can sketch on a napkin to its energetics in water versus vacuum, in one run, on a workstation.
The reaction the prize is built on
Amine-functionalized MOFs capture CO₂ the way amines always have: the nitrogen lone pair attacks the electron-poor carbon of CO₂. With one amine you get a neutral carbamic acid (R–NH–COOH); with a second amine to accept the proton you get an ion pair, an ammonium carbamate (R–NH–COO⁻ · R–NH₃⁺). Which one forms is not academic: it sets the capacity, the regeneration energy and the humidity sensitivity of a real capture material. We model the site with the simplest faithful stand-in for the appended amine, methylamine, and let Hilbeon decide.
Carbon dioxide. The central carbon is electron-poor — the target for the amine's lone pair. The two C=O double bonds make CO₂ linear and unreactive until a nucleophile reaches that carbon.
What Hilbeon finds: dry versus humid
We optimized every species at B3LYP/6-31G(d) with Hilbeon's geodesic RIC-RFO optimizer and computed the reaction energy in two environments — gas phase and, to mimic a humid pore, a C-PCM water continuum. The diagram below is the whole story.
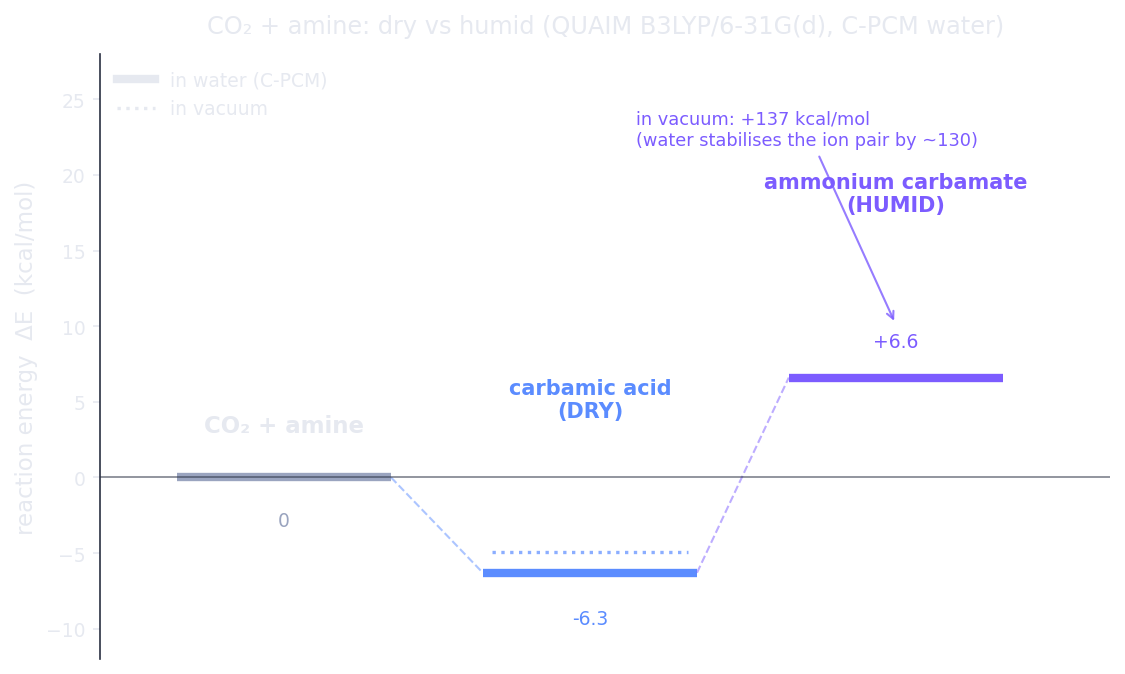
Reaction energetics (Hilbeon B3LYP/6-31G(d), gas vs C-PCM water). The neutral carbamic acid is downhill in both. The ionic ammonium carbamate is impossible in vacuum (+137 kcal/mol) but water stabilises the ion pair by ~130 kcal/mol, dropping it to +6.6 — now competitive.
Read the diagram and the chemistry explains itself. Dry: CO₂ + methylamine → N-methylcarbamic acid is favourable, −4.9 kcal/mol in the gas phase (−6.3 in C-PCM water). The neutral product forms readily. The ionic ammonium carbamate, meanwhile, is hopeless in vacuum — +137.1 kcal/mol, the brutal cost of separating a positive and a negative charge in empty space. Humid: switch on a water continuum and that same ion pair is stabilised by roughly 130 kcal/mol, collapsing to +6.6 kcal/mol — now within a hair of the carbamic acid, i.e. accessible.
That is exactly the dry-to-humid product shift reported for amine-functionalized MOFs: under dry conditions the chemisorption product is the carbamic acid, but in the presence of water vapour the equilibrium swings to the ammonium carbamate — the dry-versus-humid product shift documented experimentally for an amine-functionalized framework (J. Am. Chem. Soc. 2017; ref. below). Humidity does not just dilute the capture — it literally changes the molecule that forms. The molecular basis of that effect is visible here from first principles, water against vacuum, in a single calculation.
| Reaction | ΔE gas (kcal/mol) | ΔE in water, C-PCM (kcal/mol) |
|---|---|---|
| Dry: CO₂ + CH₃NH₂ → N-methylcarbamic acid (neutral) | −4.9 | −6.3 |
| Humid: CO₂ + 2 CH₃NH₂ → carbamate⁻ + ammonium⁺ (ion pair) | +137.1 | +6.6 |
Hilbeon B3LYP/6-31G(d) reaction energies on geodesic-RIC-RFO-optimized geometries. SMILES: CO₂ O=C=O, methylamine CN. The geometries and energies are fully deterministic and reproducible to the last digit across machines.
Who attacks whom
The two 3D structures below are the before and after of the dry reaction. Methylamine carries the reactive nitrogen lone pair; in the carbamic-acid product that nitrogen has formed a fresh N–C bond to the (now pyramidalised) carbon, with the two oxygens splitting into a C=O and a C–OH. The whole capture event is that one bond forming.
Methylamine, the model appended amine. The nitrogen lone pair is the nucleophile that reaches the CO₂ carbon.
The dry product: N-methylcarbamic acid. The amine nitrogen now bonds the carbon; CO₂'s two C=O bonds have become one C=O and one C–OH. This is the captured-CO₂ molecule.
What this is — and is not. Hilbeon models the binding-site reaction with a model amine (methylamine), not the periodic MOF framework: there is no metal node, no pore, no cooperativity between neighbouring amines here. Solvation is an implicit continuum (C-PCM), not explicit water molecules hydrogen-bonding the ions. The numbers are electronic energies (no zero-point or thermal corrections, ~0.5–1 kcal/mol), and a small D4 dispersion term is omitted as negligible for these light species. So treat the absolute kcal/mol as indicative, not benchmark. What is robust — and what reproduces the experiment — is the qualitative dry-to-humid shift: neutral carbamic acid when dry; the ionic carbamate becoming accessible only once water can pay for the charge separation.
Run the chemistry behind the headline
From a reaction you can sketch to its energetics in water versus vacuum, in one run, on a workstation — no HPC cluster. Point Hilbeon at the bond you care about.
Why a single-molecule engine still has something to say
Reticular chemistry is a triumph of materials design — building giant ordered structures from molecular bricks. But the function, capturing CO₂, is decided by a handful of atoms doing ordinary molecular chemistry. A tool that nails that local reaction — its product, its energetics, its sensitivity to water — is exactly what you want when you are tuning the amine, choosing a solvent or asking why a material loses capacity on a humid day. Hilbeon puts that answer one plain-language request away.
How to do it yourself
Everything above came from talking to the Hilbeon assistant in plain language:
build CO2 (O=C=O) and methylamine (CN) from SMILES
optimize each at B3LYP/6-31G(d)
build N-methylcarbamic acid and the ammonium-carbamate ion pair, optimize them
compute the reaction energy in gas phase and in C-PCM water
compare the dry (carbamic acid) and humid (ammonium carbamate) productsThe takeaway
The 2025 Nobel celebrates frameworks you cannot fit in a single-molecule calculation — but the CO₂ capture they are famous for comes down to one amine, one carbon and, crucially, whether water is in the room. Hilbeon reads that straight off the energetics: dry gives the neutral carbamic acid; water stabilises the ionic ammonium carbamate by ~130 kcal/mol and opens that channel, the same dry-to-humid shift measured in real amine-MOFs. Big materials, small decisive chemistry — and the small chemistry is the part you can compute on your own machine today.
Bring the quantum chemistry in-house
Start a 30-day guided pilot — every method, every core, your GPU — and run the reaction that decides your material.
References
- 2025 Nobel Prize in Chemistry (metal–organic frameworks) — NobelPrize.org
- Reticular chemistry & MOFs — H. Furukawa, K. E. Cordova, M. O'Keeffe & O. M. Yaghi, Science 2013, 341, 1230444
- The chemistry of CO₂ capture in an amine-functionalized MOF under dry and humid conditions (carbamic acid vs ammonium carbamate) — J. Am. Chem. Soc. 2017 (doi:10.1021/jacs.7b06382)
- The engine that computes it — Hilbeon