Application note
How Basic Is Your Amine? Ranking Basicity from First Principles
Basicity ADMET pKa
A basic nitrogen is one of the most common motifs in the whole drug pharmacopoeia — and it is anything but decorative. How basic that nitrogen is decides how much of your molecule walks around protonated at physiological pH 7.4, and that single fraction ripples through almost every ADMET property you care about.
Protonation state drives aqueous solubility, membrane permeability, hERG liability and lysosomal trapping. A nitrogen that is too basic locks the molecule into its charged form — great for solubility, often terrible for crossing a membrane and a notorious flag for hERG. One that is too weakly basic does the reverse. So before you commit to a scaffold, you want a number: how basic is this nitrogen, really? This post shows how Hilbeon ranks eight amine and N-heterocycle groups by their gas-phase proton affinity — the energy released when a proton lands on the basic nitrogen — and how cleanly that computed scale tracks experiment.
Why basicity is an ADMET lever, not a footnote
The basicity of an amine is usually quoted as a pKaH — the pKa of its conjugate acid, the protonated ammonium. Through the Henderson–Hasselbalch relation, pKaH and the ambient pH fix the protonated fraction directly: a nitrogen with a pKaH well above 7.4 is almost entirely cationic in the bloodstream, one well below is almost entirely neutral. That charged-vs-neutral split is the hinge. The neutral form is the one that slips through a lipid bilayer; the charged form is the one that dissolves, that can plug the hERG channel, and that gets trapped and concentrated inside acidic lysosomes. Tune the basicity and you are, whether you mean to or not, tuning all of it at once.
Underneath the aqueous pKaH sits a cleaner, more intrinsic quantity: the gas-phase proton affinity (PA), the energy to add H+ to the lone pair on the basic nitrogen. Strip away the solvent and the proton affinity is purely a property of the molecule's electronic structure — how readily that nitrogen's lone pair welcomes a proton and how well the resulting positive charge is stabilised. Electron-donating alkyl groups push density onto the nitrogen and prop up the cation; electron-poor environments do the opposite. Electronic structure is exactly what Hilbeon computes, so proton affinity is something it can rank straight from the wavefunction.
Ranking eight bases from the wavefunction
We took eight common nitrogen bases spanning the range a medicinal chemist actually meets — from weakly basic ammonia and aniline, through the alkylamine series and the aromatic N-heterocycles pyridine and imidazole, up to the strongly basic secondary amine piperidine — and for each one computed the proton affinity: the electronic energy difference between the neutral base and its protonated, positively charged form. Then we plotted Hilbeon's proton affinity against the experimental gas-phase values tabulated in the NIST Chemistry WebBook.
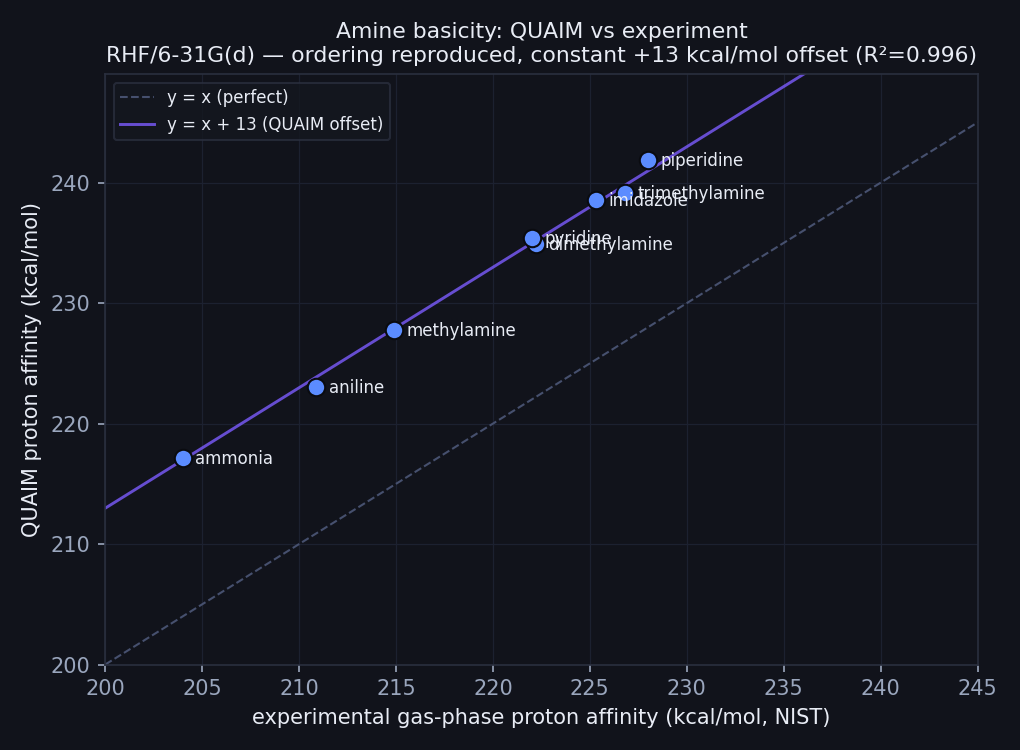
Hilbeon proton affinity vs experiment (NIST). Points sit on a line parallel to y = x: the ordering is reproduced (R² = 0.996) with a constant ≈ +13 kcal/mol offset — the net of the model-level physics a single-point RHF omits (basis/correlation, plus the missing ZPE and thermal-to-enthalpy conversion). Because it is constant it cancels in the ranking.
The result is a clean, ranked basicity scale — and a striking validation. The eight points do not just correlate with experiment; they fall on a line that runs parallel to y = x, offset bodily upward by a near-constant ≈ 13 kcal/mol. The fitted slope is 1.02 and R² = 0.996. A slope of essentially one means Hilbeon reproduces the spacing between bases, not just their order: the gap from ammonia to piperidine in the computed numbers matches the gap in the measured ones. The constant vertical shift is the only systematic discrepancy, and — crucially — because it is constant it cancels in any relative comparison. Rank two bases against each other and the offset drops out entirely.
| Amine | Hilbeon PA (kcal/mol) | Experimental PA (kcal/mol) | Offset (kcal/mol) |
|---|---|---|---|
| Ammonia | 217.1 | 204.0 | +13.1 |
| Aniline | 223.0 | 210.9 | +12.1 |
| Methylamine | 227.8 | 214.9 | +12.9 |
| Dimethylamine | 234.9 | 222.2 | +12.7 |
| Pyridine | 235.4 | 222.0 | +13.4 |
| Imidazole | 238.5 | 225.3 | +13.2 |
| Trimethylamine | 239.2 | 226.8 | +12.4 |
| Piperidine | 241.9 | 228.0 | +13.9 |
Read top to bottom, this is a basicity ladder. Ammonia anchors the bottom; each substitution that feeds electron density toward the nitrogen climbs the rungs. Aniline, whose lone pair is partly drained into the benzene ring, sits low. The alkylamines rise step by step, the aromatic heterocycles pyridine and imidazole slot in alongside the dimethyl/trimethyl region, and the secondary cyclic amine piperidine tops the list as the most basic of the set. Across all eight the offset barely moves — it ranges only from +12.1 to +13.9 kcal/mol, with a mean of 13.0 — which is precisely why the fit comes out at slope 1.02 and R² 0.996. Hilbeon never saw a single experimental number; the ordering falls straight out of the computed electron density.
Rank your own bases
From a list of SMILES to a ranked proton-affinity scale in a couple of prompts. Read the basicity of your nitrogens — before they decide your protonation state, your permeability and your hERG risk for you.
The teaching nuance: gas phase is not water
There is a classic trap hiding inside this very dataset, and it is worth pointing at directly. In the gas phase the simple alkylamines order themselves cleanly: ammonia < methylamine < dimethylamine < trimethylamine. Every methyl you add donates a little more electron density to the nitrogen, stabilises the resulting ammonium cation a little better, and raises the proton affinity — exactly the monotonic climb Hilbeon reproduces in the table above.
Drop those same amines into water, though, and that tidy order is scrambled. The aqueous basicity no longer tracks the intrinsic electronic effect, because now a second factor dominates: how well the protonated cation is solvated. A small ammonium with several N–H bonds is cradled by hydrogen bonds to water; pile on methyl groups and you strip away those N–H donors and wrap the charge in greasy, poorly-solvated alkyl, which works against basicity. The two effects pull in opposite directions and the result is a reordered, non-monotonic aqueous scale. The lesson for ADMET is blunt: a gas-phase proton affinity is a beautiful intrinsic ranking of how a nitrogen wants to behave electronically, but it is not an aqueous pKaH. For the number that actually sets your protonation fraction in plasma, you have to put the solvent back in.
What this is — and is not. These are RHF/6-31G(d) single-point energies on RDKit MMFF geometries — the structures were generated by force field and given a Hilbeon single-point electronic-structure calculation; they were not QM-reoptimized. The quantity reported is an electronic proton affinity: it carries no zero-point or thermal correction, so the absolute values run about 13 kcal/mol above experiment. That offset is the net of the model-level physics a single-point RHF leaves out — the basis-set and correlation treatment, plus the missing zero-point energy and the electronic-energy-to-298 K-enthalpy conversion that turn a raw ΔE into a measured proton affinity. The individual terms do not all push the same way, but their sum is nearly constant across the series, which is what matters here: the ranking is right — the offset cancels in any relative comparison — while the absolute number is not. And a gas-phase proton affinity is not an aqueous pKaH: the alkylamine order above is reordered in water by differential cation solvation. To turn this into solution basicity, refine with ZPE + thermal corrections (frequencies), correlation (MP2/DFT), and a solvation model (C-PCM / SMD).
Why the validation is trustworthy. A proton affinity is a difference between a neutral molecule and a charged one, the kind of comparison where a sloppy integral engine quietly leaks error. Hilbeon runs every one of these on its own integral engine (McMurchie–Davidson + Rys — no third-party integral libraries) and is numerically validated to tight tolerances and bit-identical across machines, to roughly 10−9 Ha. The numbers in the table are fully deterministic and reproducible to the last digit across machines, so the ≈ 13 kcal/mol offset you see is real, model-level physics — the basis/correlation treatment plus the missing ZPE and thermal-to-enthalpy conversion — and not numerical noise in the energies you are ranking.
How to do it yourself
Every number above came from talking to the Hilbeon assistant in plain language — no input decks, no basis-set syntax to memorize:
build pyridine from SMILES
protonate it on the basic nitrogen
run RHF/6-31G(d) on both the neutral base and the protonated cation
report the proton affinity in kcal/mol
repeat for ammonia, methylamine, dimethylamine, trimethylamine,
aniline, imidazole and piperidine
plot Hilbeon proton affinity against the NIST experimental values and fit a linePrefer scripts? Every one of those steps is also a call in Hilbeon's MCP / HTTP API, so ranking a whole library of bases is a loop in a notebook or a CI job. Point it at a scaffold whose basicity nobody has measured and read where its nitrogen lands on the same axis — well before that nitrogen quietly sets your protonation state, permeability and hERG liability for you.
The takeaway
Basicity is not a footnote on a basic nitrogen — it is the dial that sets how much of your molecule is charged at pH 7.4, and through that, its solubility, its permeability, its hERG risk and its lysosomal fate. Hilbeon ranks eight common amines and N-heterocycles by gas-phase proton affinity straight from the electron density, and the result lands on a line parallel to experiment: slope 1.02, R² 0.996, separated only by a constant ≈ 13 kcal/mol offset that is the net of the model-level physics a single-point RHF omits — basis/correlation plus the missing ZPE and thermal-to-enthalpy conversion — and, being constant, cancels in the ranking. Treat it as a relative basicity ranker, not an absolute proton-affinity oracle — remember that the gas-phase order is reshuffled in water by solvation, and refine with ZPE + thermal corrections, correlation and a solvation model when you need real aqueous pKaH — and you have a fast, first-principles way to read the basicity of a nitrogen before anyone has measured it. Point the same engine at your own series and rank the bases off the density.
Rank your own bases
Start a 30-day guided pilot — every method, every core, your GPU — and read the basicity of the nitrogens on your scaffold before you make them.
References
- Gas-phase proton affinities — NIST Chemistry WebBook (proton-affinity and gas-basicity data).
- The evaluated proton-affinity scale — E. P. L. Hunter & S. G. Lias, “Evaluated Gas Phase Basicities and Proton Affinities of Molecules,” J. Phys. Chem. Ref. Data 1998, 27, 413.
- Why basicity / ionisation state matters in ADMET — the role of pKa in permeability, solubility and hERG liability.
- From gas-phase basicity to the protonated fraction in plasma — Predicting Ionization State at pH 7.4.